J. Cent. South Univ. Technol. (2011) 18: 323-330
DOI: 10.1007/s11771-011-0699-1
Simulation of 13C NMR chemical shifts of carbinol carbon atoms using quantitative structure-spectrum relationships
DAI Yi-min(戴益民)1,2, HUANG Ke-long(黄可龙)1, LI Xun(李浔)2,
CAO Zhong(曹忠)2, ZHU Zhi-ping(朱志平)2, YANG Dao-wu(杨道武)2
1. School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China;
2. Hunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation,
Changsha University of Science and Technology, Changsha 410004, China
? Central South University Press and Springer-Verlag Berlin Heidelberg 2011
Abstract: A quantitative structure-spectrum relationship (QSSR) model was developed to simulate 13C nuclear magnetic resonance (NMR) spectra of carbinol carbon atoms for 55 alcohols. The proposed model, using multiple linear regression, contained four descriptors solely extracted from the molecular structure of compounds. The statistical results of the final model show that R2= 0.982 4 and S=0.869 8 (where R is the correlation coefficient and S is the standard deviation). To test its predictive ability, the model was further used to predict the 13C NMR spectra of the carbinol carbon atoms of other nine compounds which were not included in the developed model. The average relative errors are 0.94% and 1.70%, respectively, for the training set and the predictive set. The model is statistically significant and shows good stability for data variation as tested by the leave-one-out (LOO) cross-validation. The comparison with other approaches also reveals good performance of this method.
Key words: carbinol carbon atom; 13C nuclear magnetic resonance; chemical shift; topological indices; quantitative structure- spectroscopy relationship
1 Introduction
Nuclear magnetic resonance (NMR) has played an extremely important role in elucidating complicated structures and processes, including structural configuration [1-2], dynamic processes, reaction mechanisms and chemical equilibrium [3]. Usually, carbon-13 NMR spectrum (13C NMR) can be measured directly and a large amount of data is available. However, not all experimental structural parameters were available due to the diverse natures of the structures and reactivities [4]. It is not always possible to find reliable experimental values in practice, so the 13C NMR prediction method of the fractions is necessary and meaningful. One of the spectral simulation techniques for the identification of compounds and for the validation of their spectral assignments involves developing mathematical models that correlate the 13C chemical shifts of an atom to its structural environment [5]. Quantitative structure-spectroscopy relationships (QSSR), as a branch of quantitative structure-property/ activity relationships (QSPR/QSAR), originated from the G-P method proposed by GRANT and PAUL [6] in 1964 and L-A method by LINDEMAN and ADAMS [7] in 1971 for estimating 13C nuclear magnetic resonance spectrum of alkanes based on the addition of group contribution. Recently, many studies on 13C NMR chemical shifts simulation and prediction have been reported by artificial neural network algorithm [8-9] or multiple linear regression [10-12].
In QSPR/QSAR, it is assumed that properties or biological activities of new or untested chemicals can be inferred from the molecular structure, of which the properties or biological activities of similar compounds have already been assessed. To obtain a significant QSPR/QSAR correlation, it is crucial to find appropriate descriptors. Thus, a major and crucial task is to exactly extract the sufficient chemical information in numerical format from the molecular graph [13]. Among the numerous chemical structural descriptors, topological indices are particularly useful in compounds characterization. Topological descriptors and theoretical graph approaches in chemistry have been criticized for lacking the structural interpretation of the models. Topological indices show their vigor in QSPR/QSAR wing to the simplicity and high efficiency. In addition, some famous professional molecular modeling programs like CoMFA [14] are used in the QSPR/QSAR.
In this work, as a continuation of our earlier work [15-21], a four-parameter QSSR model was developed using a combination of topological indices with some classical molecular descriptors to simulate 13C NMR chemical shifts of carbinol carbon.
2 Experimental
2.1 Data set
A total set of 64 experimental data was selected from Ref.[22]. The number of carbons of the compounds ranged from 1 to 10, and the linear and/or branched chemical structures existed in the selected compounds. The 13C NMR chemical shift data set ranged from +49.8×10-6 to +81.8×10-6 (TMS=0). To ensure a large enough training set, 64-compound data set was randomly split into a 55-member training set and a 9-member external prediction set.
2.2 Descriptor calculation
At the molecular level, the chemical shift of NMR spectroscopy is influenced by many factors. However, the important ones are the local chemical microenvironment and the hybridization state [4]. The micro-chemical environment is related to the distribution of electron density. And the distribution of electron density is related principally to the atom electro- negativity and the bond distance. According to the principle of the nuclear magnetic resonance spectroscopy, the maximum absorption peak of 13C NMR can be expressed as
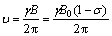
 (1)
(1)
where σ is the shield constant and B0 is the intensity of additional magnetic field. It is well known that 13C NMR chemical shifts of nucleus are related to the de-shielding effect [23]. The interpretation of NMR chemical shift values for elements other than hydrogen is known to be difficult [24]. So, the NMR spectroscopy influence factors of alcohol derivatives can be thought to be divided into two parts: inner shielding and outer shielding. Generally, the inner shielding indicates the shielding of the electron density around the atom nucleus itself. Compared with the inner shielding, the outer shielding is mainly produced by the steric effects of other atoms. However, the inner shielding and the outer shielding are combined together in some cases because of the complexity of the chemical structures. Therefore, the NMR spectroscopy influence factors sometimes do not show obvious regularities. In general, molecular structure, electronic and steric effects are the three crucial factors to determine the distribution of 13C NMR chemical shifts.
Here, a novel topological index YC is proposed. YC encodes the information of molecular mass, structure, and intermolecular interactions which are important for modeling 13C NMR chemical shifts of carbinol carbon. It is adapted from traditional distance matrix, and has equilibrium electro-negativity and relative bond as components in order to characterize atom electro- negativity and bond distance. The electro-negativity of atom A, xA, represents the ability of atoms to obtain or lose electrons when it is in a compound. The larger the electro-negativity of an atom, the stronger the ability of atom to attract electrons [25]. However, it is considered that the electro-negativity of the atom is changed in the formation of a molecule. As long as a molecule is formed, the electro-negativity of the atoms in the molecule is in the state of equilibrium, which is called the equilibrium electro-negativity.
Based on the Pauling electro-negativity, the group electro-negativity, xG, can be calculated by the method of stepwise addition [26]. It is defined as a weighted average electro-negativity of its components. The group electro-negativity of a group structural tree is illustrated in Fig.1.
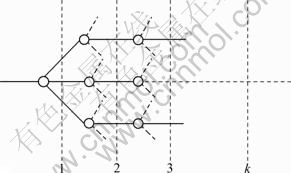
Fig.1 Plot of group structural tree
When the group is a single atom, its group electro- negativity is Pauling electro-negativity of this atom. For a group with more than two levels, all the atoms or groups attached to “anchor atom” are weighted equally, which can be expressed as follows.
The equilibrium of the first level:
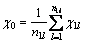
The equilibrium of the second level:

…
The equilibrium of the k-th level:
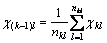
Then, group electro-negativity is defined as
 (2)
(2)
For a molecule with an equilibrium structure, the equilibrium electro-negativity of atom i is defined as
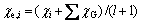 (3)
(3)
where χi is the Pauling electro-negativity of atom i, l is the total number of groups attached to i, and χG is the electro-negativity of group G that is directly connected to i.
On the other hand, the relative bond length of two adjacent vertices is used to distinguish heteroatom- containing compounds. Here, Lij is the shortest distance between vertices i and j, and is calculated by summing up the bond length between two adjacent vertices in the shortest path. If C―C bond length, LC―C=0.154 nm, is employed, then the relative bond length between vertices i and j is calculated: dij=Lij/LC―C, for example, C―O relative bond length is 0.143/0.154=0.928 6.
The distance matrix D of n atoms in a molecular skeleton graph, a symmetric matrix, can be expressed as D=[dij]n×n. In addition, vertex matrix R and equilibrium electro-negativity matrix X are defined in order to distinguish the level of branching of the molecule and the atomic species, respectively. According to these definitions, the matrix D, R and X are expressed as
 ,
, 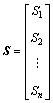 ,
, 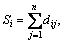
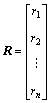 ,
, 
Taking them into account, the novel topological indices YC is defined as
 (4)
(4)
For example, the non-hydrogen skeleton of 4-Methyl-2-Pentanol is given in Fig.2 and the corresponding matrix D, R and X are given below.
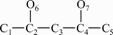
Fig.2 Non-hydrogen skeleton of 4-Methyl-2-Pentanol
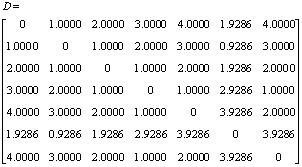 ,
,
 ,
,  ,
, 
For this molecule, topological index, YC=2.549 6.
It is worth mentioning that topological indices are the theoretical graph descriptors obtained by transforming molecular structures into corresponding molecular graph. Such transformation is performed by deleting all the carbon-hydrogen and heteroatom- hydrogen bonds in the molecular structures [27]. In this study, taking equilibrium electro-negativity of atom into account leads to successful establishment of topological index YC. It can make up the absence of hydrogen atom in a non-hydrogen skeletal graph.
In addition, the equilibrium electro-negativity of carbinol carbon is very important in determining 13C NMR chemical shifts because the equilibrium electro- negativity of the atom can effectively reflect the electron density. Accordingly, the equilibrium electro-negativity of the atom is considered to represent the information of molecular structure. The equilibrium electro-negativity of the atom will be used as the separate influence factor in the regression model.
The outer shielding is mainly concentrated on the effect of substituents. It can usually be ignored as far as four bonds are away from or directly through space [8]. Within three chemical bond distances, the influence of α, β, γ hydrogen atoms on the external magnetic field can be approximately represented by their atom numbers [28].
3 Results and discussion
3.1 Multiple regression model
To select the set of descriptors that are most relevant to the mobility of compounds, the linear models with the number of variables from 1 to 5 were built. Adding another descriptor did not significantly improve the statistics of a model, then the optimum subset size was achieved. The effects of the number of the descriptors on the correlation coefficient (R) and the standard deviation (S) are shown in Fig.3. It can be seen that the four descriptors appear to be sufficient for a successful regression model. Based on the aforementioned results and the discussion, α and β hydrogen atoms are used as the stereoscopic effect descriptors for predicting 13C NMR chemical shifts. Molecular descriptors YC, χe, NαH, NβH are given in Table 1.

Fig.3 Influence of number of descriptors on correlation coefficient (R) and standard error (S) of regression model
In this work, the experimental data set has 64 alcohols, including a set of 55 members and a set of 9 external predictions. Multiple linear regression (MLR) analysis is used to construct the QSSR model by using the four parameters, YC, χe,  and
and 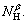 The relationship between the molecular structural descriptors and the 13C NMR chemical shift (SC) on carbinol carbon atoms is modeled by Eq.(5) as
The relationship between the molecular structural descriptors and the 13C NMR chemical shift (SC) on carbinol carbon atoms is modeled by Eq.(5) as
SC=a0+a1YC+a2χe+a3+a4 (5)
(5)
where a0 is a constant, a1, a2, a3 and a4 are the contribution coefficients of YC, χe, and 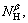 respectively. The quality of the model is determined by the standard error of estimation (ES), the prediction error sum of squares (SS) of validation, the value of Fisher statistic (F), and the cross-validated correlation coefficients (R2 and
respectively. The quality of the model is determined by the standard error of estimation (ES), the prediction error sum of squares (SS) of validation, the value of Fisher statistic (F), and the cross-validated correlation coefficients (R2 and  The results indicate that the 13C NMR chemical shifts on carbinol carbon atoms can be described by multiple linear regression:
The results indicate that the 13C NMR chemical shifts on carbinol carbon atoms can be described by multiple linear regression:
SC=-151.684 6+1.618 4YC+84.683 4χe+1.065 1+
1.715 8 (6)
where n=55, R2=0.982 4, 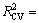 0.974 0, ES=0.869 8, SS= 37.828 3, F=697.01, p<0.000 0.
0.974 0, ES=0.869 8, SS= 37.828 3, F=697.01, p<0.000 0.
In Eq.(6), the high correlation coefficient and the low standard deviation of the model indicate that the molecular descriptors YC, χe,and are responsible for the 13C NMR chemical shifts on carbinol carbon atoms. The 13C NMR chemical shift values and deviations calculated using Eq.(6) are listed in Table 1 and shown in Fig.4(a) and Fig.5(a).
3.2 Model validation and prediction
In order to assess the predictive ability and to check the statistical significance of the developed model, leave-one-out cross-validation (LOO-CV) and external validation (EV) procedures were used [5].
3.2.1 Leave-one-out cross-validation
Validation of the models is very important for QSPR. It is quite important to measure the predictive ability and the stability of a model. The most popular validation criterion to explore the robustness of a predictive model is to analyze the influence of each individual object that configures the final equation. This procedure is known as cross-validation (CV) or internal validation by leave-one-out (LOO) [29]. The model was adopted to test the internally predictive ability for Eq.(6). The parameters of the method can play important roles in assessing the performance of models, which are SS, DS and RCV [4]:
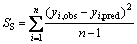 (7)
(7)
 (8)
(8)
 (9)
(9)
where n is the number of compounds included in the model, yi,obs and yi,pred are the experimental and predicted 13C NMR chemical shifts on carbinol carbon atoms of the left-out compound i, respectively and yi,avg is the average experimental 13C NMR chemical shift on carbinol carbon atoms of left-in compounds different from i. The RCV value can be considered as a measure of the predictive power of a model. Though RCV can always be artificially increased by adding more parameters, RCV decreases if a model is over-parameterized, therefore it is a more meaningful summary statistic parameter for predictive model.
The SS value being less than DS demonstrates that the model has much better predictive ability than chance as well as statistical significance [30]. Usually, the ratio of SS/DS can be used to calculate the approximate confidential interval of prediction. For a reliable model, the SS/DS ratio should be less than 0.4. If the SS/DS ratio is less than 0.1, the model is excellent. The cross-validated parameters for the proposed model are obtained as: RCV =0.986 9, SS=55.923 4, SCV =1.027 2, DS=2 147.164 4 and SS/DS=0.026 0. Cross-validation RCV value is very close to the corresponding R value and the cross-validated SCV is only slightly larger than the corresponding S value, and the corresponding results are shown in Fig.4(b). Clearly, the cross-validation demonstrates that the final model is statistically significant [31].
Table 1 Molecular descriptors and 13C NMR chemical shifts on carbinol carbon atoms of 55 alcohols
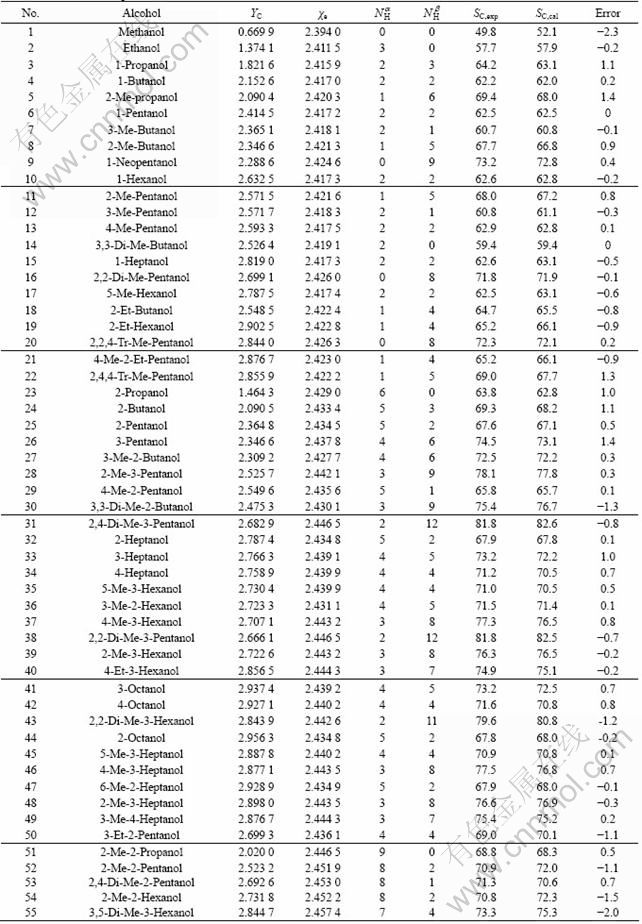

Fig.4 Plots of 13C chemical shift calculated by MLR (a) and predicted by LOO-CV (b) for total data set of 55 carbinol carbon atoms versus experimental values
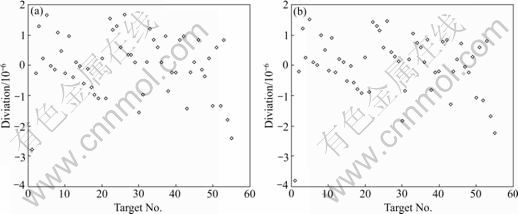
Fig.5 Diviations of 13C chemical shift: (a) Calculated values by MLR; (b) Predicted values by LOO-CV
In order to investigate the error distribution, the diviations of the calculated values by MLR and the predicted values by LOO-CV versus experimental values are plotted in Fig.5. The plotting of the calculated and predictive residuals of 13C NMR chemical shifts on carbinol carbon atoms falls, as expected, within a horizontal band centered around zero. This reveals that the deviations are randomly distributed and do not follow any kind of pattern. As shown in Fig.5, the residuals seldom exceed the standard deviation of ±2S. Therefore, no systematic rules in the errors are found, which is in agreement with the general multiple linear theory.
3.2.2 External validation
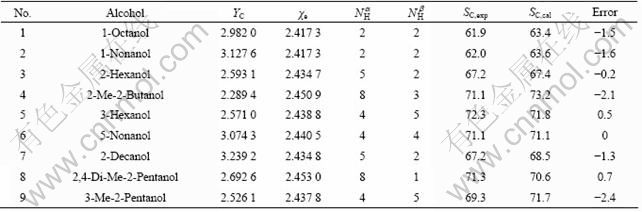
In recent years, the LOO press statistics have been used as a means of indicating predictive ability, and high values for these statistics are considered as the indicator, or even as the ultimate proof, of the high predictive power of a QSPR models [32]. However, in Ref.[33], GOLBRAIKH and TROPSHA demonstrated that a high value of LOO statistics  appears to be necessary, but not sufficient condition, for the model to have a high predictive power [33]. Therefore, these molecules have no contribution in the model development and can be used to validate the ultimate prediction ability of the resulted models. In order to further check the capability of the model, it was tested with an external set of chemicals not included in the training set. In this study, the validation data set included 9 compounds with a diverse selection of chemical structures [22]. The average relative error is 1.70% between predictive and experimental 13C NMR chemical shifts on carbinol carbon atoms, within the range of experimental error. The validated compounds molecular structural descriptors and predictive results are listed in Table 2, which indicates that their chemical shifts can be well predicted by the model.
appears to be necessary, but not sufficient condition, for the model to have a high predictive power [33]. Therefore, these molecules have no contribution in the model development and can be used to validate the ultimate prediction ability of the resulted models. In order to further check the capability of the model, it was tested with an external set of chemicals not included in the training set. In this study, the validation data set included 9 compounds with a diverse selection of chemical structures [22]. The average relative error is 1.70% between predictive and experimental 13C NMR chemical shifts on carbinol carbon atoms, within the range of experimental error. The validated compounds molecular structural descriptors and predictive results are listed in Table 2, which indicates that their chemical shifts can be well predicted by the model.
Table 2 Input parameters and predicted results for predicted set
3.3 Comparison with other approaches
JAISWAL and KHADIKAR [34] proposed the 13C NMR chemical shift model of carbinol carbon atoms including 32 alcohols using Winer (W), PI (Padmaker- Ivan) and connectivity indices (mx, mxv) of five molecular structure descriptors, the regression coefficient of the model, R=0.888 8, and the standard deviation of the model, S=3.914 9. In this four-descriptor model, only the average absolute deviation is given, which is 0.94% for 55 substances, and the correlation coefficient and standard deviation of model are superior to the aforementioned works. Furthermore, the results show that no degeneracy is present in this descriptor, while high degeneracy is present in Winer (W), PI (Padmaker- Ivan) and connectivity indices (mx, mxv). For comparison, this model is found to be far superior to JAISWAL and KHADIKAR model.
4 Conclusions
A QSSR model for 13C NMR chemical shift prediction of carbinol carbon atoms by using heuristic method based on descriptors calculated from molecular structure was developed. The squared correlation coefficient of model is 0.982 4 and the standard error of model is 0.869 8 for the training set. The stability for internal samples and prediction capacity for external samples of four-descriptor model have been tested by the leave-one-out cross validation as well as an external test. The results show that most of the predicted values of the 13C NMR chemical shifts on carbinol carbon atoms agree with the experimental values, within the range of experimental error.
References
[1] NEUVONEN H, NEUVONEN K. Correlation analysis of carbonyl carbon 13C NMR chemical shifts, IR absorption frequencies and rate coefficients of nucleophilic acyl substitutions: A novel explanation for the substituent dependence or reactivity [J]. J Chem Soc, Perkin Trans, 1999(2): 1497-1502.
[2] WITKOWSKI S, MACIEJEWSKA D, WAWER I. 13C NMR studies of conformational dynamics in 2,2,5,7,8-entamethylchroman-6-ol derivatives in solution and the solid state [J]. J Chem Soc, Perkin Trans, 2000(2): 1471-1476.
[3] W?THRICH K. The way to NMR structures of proteins [J]. Nat Struct Biol, 2001, 8: 923-925.
[4] TONG J B, LIU S L, ZHOU P, ZHANG S W, LI Z L. Quantitative structure spectroscopy relationships of carbon-13 nuclear magnetic resonance chemical shifts of steroids [J]. J Mol Graph Model, 2007, 26: 86-92.
[5] GHAVAMI R, NAJAFI A, SAJADI M, DJANNATY F. Genetic algorithm as a variable selection procedure for the simulation of 13C nuclear magnetic resonance spectra of flavonoid derivatives using multiple linear regression [J]. J Mol Graph Model, 2008, 27: 105-115.
[6] GRANT D M, PAUL E G. Carbon-13 magnetic resonance: II. Chemical shift data for the alkanes [J]. J Am Chem Soc, 1964, 86: 2984-2990.
[7] LINDEMAN L P, ADAMS J Q. Carbon-13 magnetic resonance spectrometry-chemical shifts for the paraffins through C9 [J]. Anal Chem, 1971, 43: 1245-1252.
[8] MEILER J, MAIER W, WILL M, MEUSINGER R. Using neural networks for 13C NMR chemical shift prediction-comparison with traditional methods [J]. J Magn Reson, 2002, 157: 242-252.
[9] JALALI-HERAVI M, SHAHBAZIKHAH P, ZEKAVAT B, ARDEJANI M S. Principal component analysis―ranking as a variable selection method for the simulation of 13C nuclear magnetic resonance spectra of xanthones using artificial neural networks [J]. QSAR Comb Sci, 2007, 26: 764-772.
[10] KAHRS O, BRAUNER N, GHOLAKOV G S, STATEVA R P, MARQUARDT W, SHACHAM M. Analysis and refinement of the targeted QSPR method [J]. Comput Chem Engin, 2008, 32: 1397-1410.
[11] LIANG G Z, MEI F, ZHOU Y, ZHOU P, LI Z L. Simulation of 13C nuclear magnetic resonance spectra for derivatives of bases and nucleotides [J]. Chin J Anal Chem, 2006, 34: 329-332. (in Chinese)
[12] DUCHOWICZ P R, GARRO J C M, CASTRO E A. QSPR study of the Henry’s law constant for hydrocarbons [J]. Chemometr Intell Lab Syst, 2008, 91: 133-140.
[13] POMPE M, RANDI? M. “Anticonnectivity”: A challenge for structure-property-activity studies [J]. J Chem Inf Model, 2006, 46: 2-8.
[14] CRAMER R D III, PATTERSON D E, BUNCE J D. Comparative molecular field analysis (CoMFA): 1. Effect of shape on binding of steroids to carrier proteins [J]. J Am Chem Soc, 1988, 110: 5959-5967.
[15] DAI Y M, LI X, CAO Z, YANG D W, HANG K L. Modeling flash point scale of hydrocarbon by novel topological electro-negativity indices [J]. CIESC Journal, 2009, 60(10): 2420-2425. (in Chinese)
[16] DAI Y M, LI X, LIANG B, YANG D W, CAO Z, HANG K L. Quantitative relationship between 13C nuclear magnetic resonance chemical shift and structural parameters of acyclic alcohol [J]. Chin J Anal Chem, 2009, 37(12): 1754-1758. (in Chinese)
[17] DAI Y M, DENG X Q, YANG D W, ZHOU H M. A quantitative structure-spectrum relationship study of 13C NMR chemical shifts of halogenated methane [J]. Chin J Magn Reson, 2008, 25(1): 110-116. (in Chinese)
[18] DAI Y M, WEN S N, NIE C M, LI Z H. A novel quantum topological index and predicting physical-chemical properties of the lanthanide [J]. Chin J Inorg Chem, 2005, 21(8): 1015-1019. (in Chinese)
[19] LI Z H, DAI Y M, WEN S N, NIE C M, ZHOU C Y. Relationship between atom valence shell electron quantum topological indices and electro-negativity of elements [J]. Acta Chim Sinica, 2005, 63(14): 1348-1356. (in Chinese)
[20] ZHOU C Y, NIE C M, LI S, LI Z H. A novel semi-empirical topological descriptor Nt and the application to study on QSPR/QSAR [J]. J Comput Chem, 2007, 28: 2413-2423.
[21] ZHOU C Y, CHU X, NIE C M. Predicting thermodynamic properties with a novel semi-empirical topological descriptor and path numbers [J]. J Phys Chem B, 2007, 111: 10174-10179.
[22] Sadtler research laboratories division of Bio-Rad laboratories, INC. Sadtler standard carbon-13 NMR spectra [M]. USA, 1980.
[23] TONG J B, LIU S L, ZHOU P, ZHANG S W, LI S S. Prediction of 31P nuclear magnetic resonance chemical shifts for phosphines [J]. Spectrochimica Acta: Part A, 2007, 67: 837-846.
[24] BOSQUE R, SALES J. A QSPR study of the 31P NMR chemical shifts of phosphines [J]. J Chem Inf Comput Sci, 2000, 41: 225-232.
[25] LU C H, GUO W M, HU X F, WANG Y, YIN C S. A Lu index for QSAR/QSPR studies [J]. Chem Phys Lett, 2006, 417: 11-15.
[26] NIE C M. Group electro-negativity [J]. J Wuhan Univ: Nat Sci Ed, 2000, 46(2): 176-180. (in Chinese)
[27] LIU S S, XIA Z N, YU B M, LI Z L. Atomic electronegative distance vector of acyclic alcohol and chemical shifts simulation of 13C NMR spectroscopy [J]. Chin J Magn Reson, 1999, 16(5): 429-440. (in Chinese)
[28] LIU H X, YAO X J, LIU M C, HU Z D, FAN B. Prediction of gas-phase reduce ion mobility constants (K0) based on the multiple linear regression and projection pursuit regression [J]. Talanta, 2007, 71: 258-263.
[29] JOHNSON R A, WICHERN D W. Applied multivariate statistical analysis [M]. Upper Saddle River, NJ: Prentice-Hall, 1988: 130-139.
[30] AGRAWAL V K, BANO S, KHADIKAR P V. QSAR study on 5-Lipoxygenase inhibitors using distance-based topological indices [J]. Bioorg Med Chem, 2003, 11: 5519-5527.
[31] LIU F P, LIANG Y Z, CAO C Z, ZHOU N. QSPR study of GC retention indices for saturated esters on seven stationary phases based on novel topological indices [J]. Talanta, 2007, 72: 1307-1315.
[32] GAO S, CAO C Z. Extending bond orbital-connection matrix method to the QSPR study of alkylbenzenes: Some thermochemical properties [J]. J Mol Struct (THEOCHEM), 2006, 778: 5-13.
[33] GOLBRAIKH A, TROPSHA A. Beware of q2 [J]. J Mol Graph Model, 2002, 20: 269-276.
[34] JAISWAL M, KHADIKAR P. QSAR study on 13C NMR chemical shifts on carbinol carbon atoms [J]. Bioorg Med Chem, 2004, 12: 1793-1798.
(Edited by YANG Bing)
Foundation item: Projects(20775010, 21075011) supported by the National Natural Science Foundation of China; Project(2008AA05Z405) supported by the National High-tech Research and Development Program of China; Project(09JJ3016) supported by the Natural Science Foundation of Hunan Province, China; Project(09C066) supported by the Scientific Research Fund of Hunan Provincial Education Department, China; Project(2010CL01) supported by the Foundation of Hunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, China
Received date: 2010-03-03; Accepted date: 2010-10-13
Corresponding author: HUANG Ke-long, Professor, PhD; Tel: +86-731-88879850; E-mail: klhuang@mail.csu.edu.cn

