
Diversity of microbial community at acid mine drainages from Dachang metals-rich mine, China
ZHOU Zhi-jun(周智君)1, 2, YIN Hua-qun(尹华群)1, 2, LIU Yi(刘 毅)1, 2,
XIE Ming(谢 明)1, 2, QIU Guan-zhou(邱冠周)1, 2, LIU Xue-duan(刘学端)1, 2
1. School of Minerals Processing and Bioengineering, Central South University, Changsha 410083, China;
2. Key Laboratory of Biometallurgy of Ministry of Education, Central South University, Changsha 410083, China
Received 20 May 2009; accepted 23 July 2009
Abstract: Two acid mine drainage (AMD) samples TS and WK, which were from the Dachang metals-rich mine in Guangxi province, China, were studied using PCR-based cloning approach. A total of 44 operational taxonomic units (OTUs) were obtained from the two AMD samples. However, only three OTUs (GXDC-9, GXDC-19 and GXDC-50) detected in sample TS can also be observed in sample WK. Phylogenetic analysis revealed that the bacteria in the two samples fell into four putative divisions, which were Nitrospira, Alphaproteobacteria, Gamaproteobacteria, and Acidobacteria. Organisms of genuses Acidithiobacillus and Leptospirillum, which were in gamaproteobacteria class and Nitrospira family, were dominant in two samples, respectively. In sample TS, which was characterized by low pH, high sulfate, high iron, and high arsenide, two species (Acidithiobacillus ferrooxidans and Leptospirillum ferrooxidans) constituted 98.22% of the entire microbial community. Compared with sample TS, the microbial community in sample WK was more diversified according to the observation. Interestedly, the Legionella species, which was rarely observed in the low-pH environment, was detected in sample WK. This work helps us to further understand the diversity of microbial community living in extreme acid mine drainages with unique geochemistry and the tolerance capability of acidophiles to heavy metal.
Key words: microbial community; acid mine drainage (AMD); tolerance capability; PCR-based cloning approach
1 Introduction
Acid mine drainage (AMD) systems and the inhabiting microbial communities have aroused interest of researchers worldwide because of the unique environmental conditions of low pH, high concentration of metal iron, and so on[1-3]. Due to the ability of the microorganisms to survive, grow and reproduce in such harsh environment as well as the potential for these microorganisms to be utilized in extraction of metal Cu, Au, from low grade ores[4], study of the microbial community composition in AMD can provide insights into the relative abundance of the microorganisms and help to reveal different roles that microorganisms play in community. Previous studies of microbial diversity showed that the microbial community composition is largely bound to geochemical parameters such as pH and metal ion concentration[5-6]. For example, JOHNSON and HALLBERG[7] systematically analyzed the physicochemical and microbiological characteristics of acid mine drainage from various sites worldwide, and found that the relative abundances of Fe-oxidizers, S-oxidizers and other Herotrophic acidophiles were different at dissimilar sites, as well as microorganism community composition. Even if the geography and geochemistry properties in different sites showed some similarity, there were some significant differences at microbiological level[8].
Restriction fragment length polymorphism (RFLP), also known as amplified ribosomal DNA restriction analysis (ARDRA) is a very useful tool to study microbial diversity that relies on DNA polymorphisms [9]. Using RFLP method, YIN et al[10] successfully analyzed the microbial communities in three different mine drainages. In order to better understand the compositions and structures of microorganisms in different AMD environments, two AMD samples from the Dachang metals-rich mine located at Guangxi province of China were studied. The Dachang mine has been mined for metals for more than 20 years due to its high concentration of sulfur and arsenide. However, the microbial communities in AMD samples from the Dachang mine have not been investigated yet. The aims of this study were to investigate the bacterial diversity at these two sites and to further expand the global view of microbial diversity in AMD environments.
2 Materials and methods
2.1 Site description and sample collection
Samples TS and WK were collected from Teishu reservoir and Weikang reservoir at Dachang metals-rich mine, Guangxi province, China. This mine has been mined for metals for more than 20 years, which has resulted in a large amount of acid mine drainages since some sulfide mineral surface area and the reactive ore are exposed to oxygen and water. The two samples TS and UK represented the two kinds of different acid mine drainage located at the Dachang metals-rich mine. Teishu is the first kind of reservoir discarded for many years in the mine district. Weikang was another kind of reservoir used for collecting the acid mine drainage formed during oxidation of low-grade mine.
Water samples for molecular analysis of microbial populations were collected in August 2006. About 20 L water from each site was collected. The water was then filtered through a sterile 0.22 μm nucleopore filter (Durapore, Millipore, Bedford, MA, USA). The sediments on the filter were then immediately transferred to a tube and stored at -20 ℃ for further molecular analysis.
2.2 Chemical analysis of water sample
The concentrations of elements were measured by inductively coupled plasma-atomic emission spectroscopy (ICP-AES; Baird Plasma Spectrovac PS-6 (N+1)). Twenty-four elements were tested in both water samples.
2.3 DNA extraction and purification
The bulk communities DNA of two samples were extracted from 5 g sediments according to the protocol described by ZHOU et al[11]. The crude DNA was further purified by agarose gel electrophoresis (1% agarose) and the Wizard DNA Clean-Up Kit (Promega, Madison, Wis.). DNA quantity was determined by spectrophotometry (Nanodrop Technologies, Rockland, DE). All DNA were stored at -20 ℃ until being used.
2.4 PCR and fractionation of 16S rRNA genes
The 16S rRNA genes of the microbial community were amplified. The reaction system contained 100 ng DNA template, 1xPCR buffer (10 mmol/L Tris-HCl (pH 8.3), 50 mmol/L KCl, 2 mmol/L MgCl2, and 0.001% (w/v) gelatin), 2 mmol/L dNTPs, 5 pmol/L each of the forward and reverse primers, and 2.5 U of AmpliTaq Gold (Perkin Elmer). The final volume of 50 μL was adjusted with distilled water. The reverse primer was the universal 1387R (5′-GGGCGGWGTGTACAAGGC-3′) and the forward primer was the universal 63F (5′-CAG- GCCTAACACATGCAAGTC-3′)[12]. The thermal cycling protocol was an initial denaturation at 94 ℃ for 5 min, followed by 30 cycles of 94 ℃ for 45 s, 55 ℃ for 45 s, and 72 ℃ for 90 s, and a final extension step under 72 ℃ for 7 min. Products from the amplification reactions of expected size (approximately 1.3 kbp) were excised from 1% regular agarose gels and purified with Wizard PCR Clean-Up system (Promega) under the manufacturer’s instructions.
2.5 Cloning and RFLP analysis of 16S rRNA genes
The purified PCR products were cloned into the vector PCR 2.1 TOPO (Invitrogen) and transformed by Escherichia coli TOP-10F competent cells according to the manufacturers’ instructions (Invitrogen, Carlsbad, Calif.). After growing overnight on plates with ampicillin (100 mg/mL) and X-gal (15 mg/mL), putative positive clones were identified based on the blue-white screening. White colonies from each library were randomly selected and the inserts were re-amplified with the vector primers M13F and M13R[13]. The products were then digested with 1 U restriction endonucleases HinPI and MspI each mixed in 1x NEB buffer (New England Biolabs, Beverly, Mass.) overnight at 37 ℃. The restriction fragments were separated by gel electrophoresis in 3.0% agarose with ethidium bromide staining and observed on UV illumination. The RFLP banding patterns were analyzed and clustered with Molecular Analyst 1.6 software (Applied Math, Kortrijk, Belgium) by using the unweighted pair group method with arithmetic averages and the Jaccard algorithm. At last, the representative clones were selected for nucleotide sequence determination.
2.6 DNA sequencing and phylogenetic analysis
To understand the phylogenetic diversity in both sites, all clones with unique RFLP banding patterns were partially sequenced. Sequence identification was estimated initially by BLASTN facility from the National Center for Biotechnology Information (http://www.ncbi. nlm.nih.gov/). Before constructing the phylogenetic tree, these 16S rRNA sequences in NCBI databases affiliated with the acidophiles were chosen as references and these acidophiles were often reported in AMD. These 16S rRNA sequences (including the references sequences and researched sequences) were then alignmented using CLUSTAL W (with the following parameters: Gap opening penalty 5.00 and gap extension penalty 0.05) [14]. The final 16S rRNA phylogenetic tree was constructed with Mega III software [15]. Pairwise distances were calculated with the distance only option and the phylogeny tree was constructed using the Neighbor-Joining method (bootstrap test of the phylogeny: 500 replicates, seed 70 189) [16].
2.7 Statistical methods
The diversity index (H) was calculated as follows: H=-∑(pi)(lg2pi), where pi is the proportion of the RFLP banding pattern. At the same time, we also performed a rarefaction analysis to check if the clone number is sufficient to detect community diversity. The relationship of the abundances of operational taxonomic units(OTUs) to the ranks of the corresponding OTUs was fitted with a nonlinear statistical model (SigmaPlot v. 8.0):
y=a[1-exp(-bx)]
where y is the abundance of an OTU, x is the rank of the corresponding OTU, and a and b are regression parameters[17].
2.8 Nucleotide sequence accession numbers
All of the sequences described in this study have been submitted to GenBank under accession numbers from EU250215 to EU250265.
3 Results
3.1 Geochemical properties of two sites
The temperature of two sites was the same of 25 ℃, but the pH values were different. The pH was 1.8 in Teishu reservoir and 2.5 in Weikang reservoir (Table 1).
Table 1 Geochemical properties and diversity index at two sites, TS and WK (mg/L)
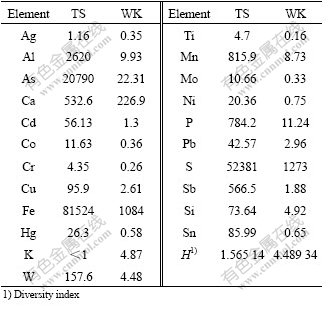
The concentrations of the elements were significantly different at the two sites, according the ICP-AES analysis. Sample TS was distinguished by the unique geochemical characteristics such as high sulfate concentration (52 381 mg/L), high iron concentration (81 524 mg/L), and especially very high concentration of arsenide (20 790 mg/L). The concentrations of these elements (sulfate, iron and arsenide) were respectively 41 and 75, 932 times higher than those of the WK sample (Table 1). Compared with the TS sample, only the concentration of element potassium in site WK (4.87 mg/L) was a little higher than that of TS sample (<1 mg/L).
3.2 RFLP analysis of 16S rRNA gene clone libraries
The PCR products of 16S rRNA (1.3 kb) gene with the expected size were successfully amplified from community genomic DNA from the samples. After T-A cloning, a total of 202 16S rRNA positive colones (112 from TK and 90 from WK) were recovered from two samples, and then, these clones were screened by RFLP analysis. The rarefaction curves suggested that the number of OTUs at all sites was close to the asymptotic level (Fig.1). RFLP analysis revealed extensive diversity of 16S rRNA genes of these samples.
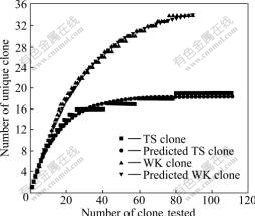
Fig.1 Evaluation of representation of clones obtained from samples TS and WK by rarefaction analysis
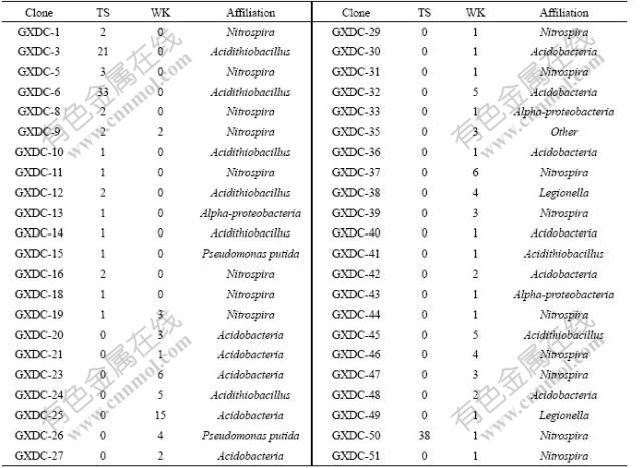
Sixteen and thirty patterns of 16S rRNA gene at TS and WK were detected, respectively. Only three patterns (GXDC-9, GXDC-19 and GXDC-50) detected in sample TS can also be detected in sample WK. In the sample TS, two RFLP patterns (GXDC-6 and GXDC-50) represented 29.5% and 33.9% among the total clone populations, respectively. In the sample WK, the dominant RFLP patterns GXDC-24 represented 16.7% of the total clone populations (Table 2). The diversity indexes calculated showed that the diversity index of sample WK (H=4.489 34) was three times higher than sample TS (H=1.565 14).
Table 2 Distribution and affiliation of clones in 16s rDNA libraries
3.3 Phylogenetic analysis
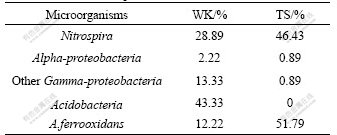
Phylogenetic tree was established with a bootstrap neighbor-joining method. According to the phylogenetic analysis, all 16S rRNA gene clone sequences fell into four putative phylogenetic classes: Alpha- and Gamma- Proteobacteria, Acidobacteria and Nitrospira (Fig.2). In sample WK, four classes were all detected. But in sample TS, only three classes were observed, which were Alpha-Proteobacteria (0.89%), Gamma-Proteobacteria (52.68%) (composed of Acidithiobacillus ferrooxidans) and Nitrospira (46.43%) (Table 3).
According to clone library analysis, the composition of microbial community in sample TS was much simpler than that of WK. Nitrospira and Gamma-Proteobacteria were the most dominant microorganisms in the phylogenetic tree based on the 16S rRNA gene sequence. Nearly all microorganisms affiliated with the Nitrospira were composed of Leptospirillum ferrooxidans except for clone GXDC-19 (Fig.2). There are eight OTUs associated with Leptospirillum ferrooxidans. Gamma-Proteobacteria was another dominant microorganism according to the phylogenetic analysis. In this class, most of the OTUs (45.54% of all clones in TS) were affiliated with Acidithiobacillus ferrooxidans except for GXDC-15, which was associated with Pseudomonas putida (0.89% of all clones at TS) (Table 3). Moreover, Acidiphilium, which was affiliated with Alpha-Proteobacteria, was also detected, but only 0.89% of the clones were associated with this class.
In sample WK, Acidobacteria was the most dominant group based on the phylogenetic analysis (43.33% of all clones), but it was not detected in sample TS. Nitrospira was the second larger population of the microbial community (28.89% of all clones) (Table 3). Compared with sample TS, the microorganisms affiliated with Nitrospira include Leptospirillum ferrooxidans, other than Leptospirillum ferriphium (Fig.2). Gamma- Proteobacteria was another dominant group, which was comprised by Acidithiobacillus ferrooxidans (17.4% of all clones) and Legionella sp. (6% of all clones). Moreover, two sequences (GXDC-33 and GXDC-43) comprising 2.22% of the entire microbial communities at WK were affiliated with Alpha-Proteobacteria.
4 Discussion
Currently, AMD environments have been set as model systems for analysis of biogeochemical interactions and microbial community structure and function[18]. Every AMD system has certain microbial niches for variations in environmental data[19-20]. In this work, culture-independent molecular methods (PCR-RFLP) were employed, including 16S rDNA clone library analysis and sequence determination. Microbial community structures in two AMD samples (TS and WK) from Dachang metals-rich mine in Guangxi province of China have been identified. According to RFLP analysis, there were 44 OTUs detected in two samples. All these 44 OTUS fell into four putative classes, which were Alpha-Proteobacteria, Gamma-Proteobacteria, Acido- bacteria and Nitrospira.
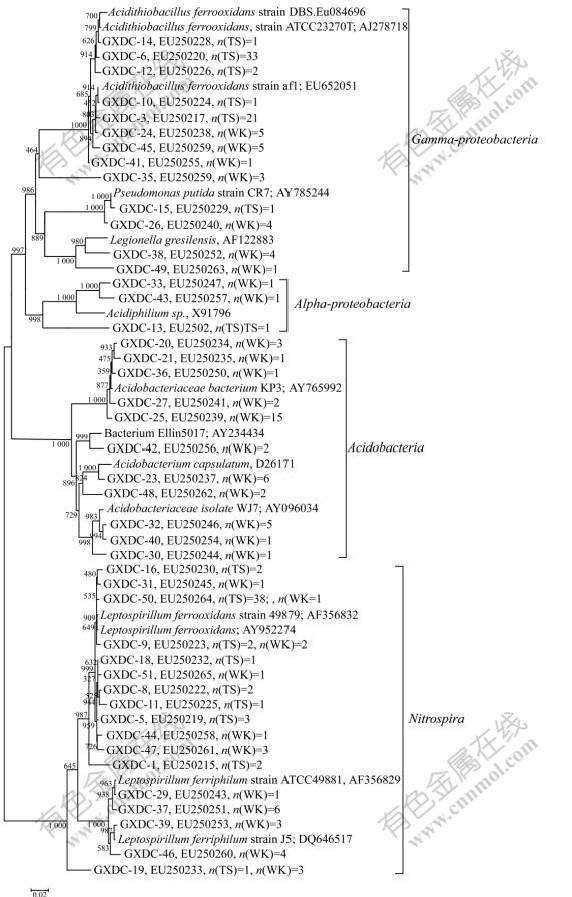
Fig.2 Phylogenetic analysis of recovered 16S rRNA gene sequence (n is number of clones)
Table 3 Microbial composition found at two sites
The results of the present study indicated that the microbial diversity level in sample TS was much lower compared with that in sample WK. The difference may be attributed to the divergence of ion concentrations at the similar temperature. Iron, sulfur, especially arsenic concentrations in sample TS were much higher than those in sample WK. It has been shown that arsenic is toxic to most typical acidophilic thermophiles used for bioleaching, either arsenate [As(Ⅴ)] or arsenite [As(Ⅲ)] [21]. The resistance of As has been reported in Acidiphilium multivorum, A.ferrooxidans, Leptospirillum, ‘Ferroplasma acidarmanus’ and Metallosphaera sedula [22-26]. Of all above microorganisms, A.ferrooxidans and Leptospirillum showed the best resistance to As(Ⅲ) (6 300 mg/L and 4 500 mg/L, respectively)[23, 26]. This might be the reason that the majority of bacteria (98.22%) in the sample TS were related to Acidithiobacillus ferrooxidans and Leptospirillum ferrooxidans. Moreover, previous studies indicated that some microorganisms affiliated with Gallionella ferruginea and Thiomonas were also often detected at the AMD with higher concentration of arsenic [26-27]. These microorganisms efficiently remove As(Ⅲ) and As(Ⅴ) in water[27]. However, these bacteria were not observed in the TS site, where the concentration of As was higher. In sample TS, Acidithiobacillus ferrooxidans and Leptospirillum ferrooxidans were two dominant microorganisms. They might play a more important ecological role in environments such as TS, compared with Gallionella ferruginea and Thiomonas.
Legionella species, a facultative intracellular Gram- negative microorganism[28-29], was frequently detected in aquatic environments and was well known for their role as the agent of Legionnaires’ disease. However, their occurrence in extremely acidic AMD environments has only been reported by KATHY et al [30]. They found four known Legionella species by investigating a predominately eukaryotic algal mat community in a pH 2.7 geothermal stream in Yellowstone National Park[30]. In the present study, Legionella species was also detected and constituted the third dominant microorganism in sample WK where the pH was 2.5. This suggested that the bacteria might be useful in the process of bioleaching or the shaping of AMD. But the actual role of this microorganism still needs to be further confirmed. Generally, isolation was necessary to define their ecophysiological roles and characteristics. But by far, there were no pure cultures of Legionella species that can grow at low-pH environments.
In the present study, the microbial diversity of AMD sites in Guangxi province was investigated and the potential relationship between geochemistry and microbial community composition was also studied. There are some interesting phenomenons such as the tolerance capability of acidophiles to heavy metal, especially as might need further clarification. From our results it seems that the geochemistries of the mine waters have an important effect on determining the composition of microbial communities.
5 Conclusions
1) The microbial diversity level in sample TS is much lower compared with that in sample WK.
2) According to clone library analysis, the composition of microbial community in sample TS is much simpler than that in sample WK. Nitrospira and Gamma-Proteobacteria are the most dominant microorganisms in the phylogenetic tree based on 16S rRNA gene sequence. In sample WK, Acidobacteria is the most dominant group based on the phylogenetic analysis (43.33% of all clones), and Nitrospira is the second larger population of the microbial community (28.89% of all clones).
3) The difference may be attributed to the divergence of ion concentrations at the same temperature. Iron, sulfur, especially arsenic concentrations in sample TS are much higher than those in sample WK. It seems that the geochemistry of the mine waters have an important effect on determining the composition of microbial communities.
References
[1] BAKER B J, BANFIELD J F. Microbial communities in acid mine drainage [J]. FEMS Microbiology Ecology, 2003, 44: 139-152.
[2] L?PEZ-ARCHILLA A I, MARIN I, AMILS R. Microbial community composition and ecology of an acidic aquatic environment: The Tinto River, Spain [J]. Microbial Ecology, 2001, 41: 20-35.
[3] SCHRENK M O, EDWARDS K J, GOODMAN R M, HAMERS R J, BANFIELD J F. Distribution of Thiobacillus ferrooxidans and Leptospirillum ferrooxidans: Implications for generation of acid mine drainage [J]. Science, 1998, 279: 1519-1522.
[4] GONZALEZ-TORIL E, LLOBET-BROSSA E, CASAMAYOR E O, AMANN R, AMILS R. Microbial ecology of an extreme acidic environment: The Tinto River [J]. Applied and Environmental Microbiology, 2001, 69: 4853-4865.
[5] JOHNSON D B, HALLBERG K B. The microbiology of acidic mine waters [J]. Research in Microbiology, 2003, 154: 466-473.
[6] TUFFIN IMARLA, HECTOR S B, DEANE S M, RAWLINGS D E. Resistance determinants of a highly arsenic-resistant strain of Leptospirillum ferriphilum isolated from a commercial biooxidation tank [J]. Appl Environ Microbiol, 2006, 72: 2247-2253.
[7] JOHNSON D B, HALLBERG K B. The microbiology of acidic mine waters [J]. Research in Microbiology, 2003, 154(7): 466-473.
[8] L?PEZ-ARCHILLA A I, MARIN I, AMILS R. A comparative ecological study of two acidic rivers in southwestern spain [J]. Microbial Ecology,, 1999, 38(2): 146-156.
[9] KIRK J L, BEAUDETTE L A, HART M, MOUTOGCIS P, KLIRONOMOS J N, LEE H, TREVORS J T. Methods of studying soil microbial diversity [J]. Journal of Microbiological Methods, 2004, 58: 169-188.
[10] YIN Hua-qun, QIU Guan-zhou, WANG Dian-zuo, CAO Lin-hui, DAI Zhi-min, WANG Jie-wei, LIU Xue-duan. Comparison of microbial communities in three mine drainages and their bioleaching efficiencies to chalcopyrite [J]. Journal of Central South University of Technology, 2007, 14(4): 460-466.
[11] ZHOU J Z, BRUNS M A, JAMES M T. DNA recovery from soils of diverse composition [J]. Appl Environ Microbiol, 1996, 62: 316-322.
[12] MARCHESI J R, SATO T, WEIGHTMAN A J, MARTIN T A, FRY J C, HIOM S J, DYMOCK D, WADE W G. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA [J]. Appl Environ Microbiol, 1998, 64: 795-799.
[13] TOKURA M, CHAGAN I, USHIDA, KOJIMA Y. Phylogenetic study of methanogens associated with rumen ciliates [J]. Current Microbiology, 1999, 39: 123-128.
[14] LARKIN M A, BLACKSHIELDS G, BROWN N P, CHENNA R, MCGETTIGAN P A, MCWILLIAM H, VALENTIN F, WALLACE I M, WILM A, LOPEZ R, THOMPSON J D, GIBSON T J, HIGGINS D G. Clustal W and Clustal X version 2.0 [J]. Bioinformatics, 2007, 23: 2947-2948.
[15] KUMAR S, TAMURA K, NEI M. MEGA3: Integrated software for molecular evolutionary genetics analysis and sequence alignment [J]. Briefings in Bioinformatics, 2004, 5: 150-163.
[16] SAITOU N, NEI M. The neighbor-joining method: A new method for reconstructing phylogenetic trees [J]. Molecular Biology and Evolution, 1987, 4: 406-425.
[17] ZHOU J Z, XIA B C, HUANG H S, ANTHONY V P, TIEDJE J M. Microbial diversity and heterogeneity in sandy subsurface soils [J]. Applied and Environmental Microbiology, 2004, 70: 1723-1734.
[18] HALLBERG K B, JOHNSON D B. Novel acidophiles isolated from moderately acidic mine drainage waters [J]. Hydrometallurgy, 2003, 71: 139-148.
[19] FERIS K, RAMSEY P, FRAZAR C, MOORE J N, GANNON J E, HOLBERT W E. Differences in hyporheic-zone microbial community structure along a heavy-metal contamination gradient [J]. Appl Environ Microbiol, 2003, 69: 5563-5573.
[20] ROHWERDER T, GEHRKE T, KINZLER K, SAND W. Bioleaching review (Part A): Progress in bioleaching: Fundamentals and mechanisms of bacterial metal sulfide oxidation [J]. Appl Environ Microbiol, 2003: 63, 239-248.
[21] MARK D, AUSTIN B C, KOPPINEEDI P R, PHILIP L B. Growth in sulfidic mineral environments, metal resistance mechanisms in acidophilic micro-organisms [J]. Microbiology, 2003, 149: 1959-1970.
[22] GIHRING T M, BOND P L, PETERS S C, BANFIELD J F. Arsenic resistance in the archaeon Ferroplasma acidarmanus, new insights into the structure and evolution of the ars genes [J]. Extremophiles, 2003, 7: 123-130.
[23] HARVEY P I, CRUNDWELL F K. The effect of As(Ⅲ) on the growth of Thiobacillus ferrooxidans in an electrolytic cell under controlled redox potential [J]. Min Eng, 1996, 9: 1059-1068.
[24] HUBER G, SPINNLER C, GAMBACORTA A, STETTER K O. Metallosphaera sedula gen. and sp. nov. represents a new genus of aerobic, metal-mobilizing, thermoacidophilic archaebacteria [J]. Syst Appl Microbiol, 1989, 12: 38-47.
[25] SUZUKI K, WAKAO N, SAKURAI Y, KIMURA T, SAKKA K, OHMIYA K. Transformation of Escherichia coli with a large plasmid of Acidiphilium multivorum AIU 301 encoding arsenic resistance [J]. Appl Environ Microbiol, 1997, 63: 2089-2091.
[26] BRUNET F B, DICTOR M C, GARRIDO F, CROUZET C, MORIN D, DEKEYSER K, CLARENS M, BARANGER P. An arsenic(Ⅲ)-oxidizing bacterial population: Selection, characterization, and performance in reactors [J]. Journal of Applied Microbiology, 2002, 93: 656-667.
[27] BRUNEEL O, DURAN R, CASIOT C, ELBAZ-POULICHET F, PERSONN? J C. Diversity of microorganisms in Fe-As-rich acid mine drainage waters of Carnoulès, France [J]. Appl Environ Microbiol, 2006, 72: 551-556.
[28] ATLAS R M. Legionella: From environmental habitats to disease pathology, detection and control [J]. Environ Microbiol, 1999, 1: 283-293.
[29] NWACHUKU N, GERBA C P. Health effects of Acanthamoeba spp. and its potential for waterborne transmission [J]. Rev Environ Contam Toxicol, 2004, 180: 93-131.
[30] KATHY B S, JOAN M H, MICHAEL J. Legionella species diversity in an acidic biofilm community in yellowstone national park [J]. Appl Environ Microbiol, 2005, 71: 507-511.
Foundation item: Projects(50321402, 30428014, 50621063) supported by the National Natural Science Foundation of China; Project(2004CB619201) supported by the National Basic Research Program of China
Corresponding author: LIU Xue-duan; Tel: +86-731-88830546; Fax: +86-731-88710804; E-mail: xueduanliu@yahoo.com
DOI: 10.1016/S1003-6326(09)60263-6
(Edited by LI Xiang-qun)

