J. Cent. South Univ. Technol. (2011) 18: 1930-1939
DOI: 10.1007/s11771-011-0925-x
16s rDNA based microbial diversity analysis of
eleven acid mine drainages obtained from three Chinese copper mines
XIE Jian-ping(л��ƽ)1, 2, 3, JIANG Hong-chen(�����)4, LIU Xin-xing(������)1, 3,
LIU Xue-duan(��ѧ��)1, 3, ZHOU Ji-zhong(�ܼ���)2, QIU Guan-zhou(�����)1, 3
1. School of Resources Processing and Bioengineering, Central South University, Changsha 410083, China;
2. Institute for Environmental Genomics, University of Oklahoma, Norman, Oklahoma, 73072, USA;
3. Key Laboratory of Biometallurgy of Ministry of Education, Central South University, Changsha 410083, China;
4. Geomicrobiology Laboratory, State Key Laboratory of Geological Processes and Mineral Resources,
School of Earth Sciences and Resources, China University of Geosciences, Beijing 100083, China
? Central South University Press and Springer-Verlag Berlin Heidelberg 2011
Abstract: Eleven acid mine drainage (AMD) samples were obtained from southeast of China for the analysis of the microbial communities diversity, and the relationship with geochemical variables and spatial distance by using a culture-independent 16S rDNA gene phylogenetic analysis approach and multivariate analysis respectively. The principle component analysis (PCA) of geochemical variables shows that eleven AMDs can be clustered into two groups, relative high and low metal rich (RHMR and RLMR) AMDs. Total 1 691 clone sequences are obtained and the detrended correspondence analysis (DCA) of operational taxonomic units (OTUs) shows that, ��-Proteobacteria, Acidobacteria, Actinobacteria, Cyanobacteria, Firmicutes and Nitrospirae are dominant species in RHMR AMDs. In contrast, ��-Proteobacteria, ��-Proteobacteria, Planctomycetes and Bacteriodetes are dominant species in RLMR AMD. Results also show that high-abundance putative iron-oxidizing and only putative sulfur-oxidizing microorganisms are found in RHMR AMD. Multivariate analysis shows that both geochemical variables (r=0.429 3, P=0.037 7) and spatial distance (r=0.321 3, P=0.018 1) are significantly positively correlated with microbial community and pH, Mg, Fe, S, Cu and Ca are key geochemistry factors in shaping microbial community. Variance partitioning analysis shows that geochemical variables and spatial distance can explain most (92%) of the variation.
Key words: acid mine drainage; community structure; 16S rDNA gene; geochemical variables; spatial distance
1 Introduction
Acid mine drainage (AMD) with low pH value and high concentration of metals and non-metallic ions is formed when minerals (principally sulfides) are exposed to air, water, and microorganisms by natural weathering or human activities [1]. Nowadays, it has become a worldwide environmental problem since it has harmful consequences for diatoms [2], protozoans [3], aquatic invertebrates [4], piscivorous birds in freshwater lakes [5], groundwater, rivers, streams [6] and surrounding vegetation [7]. However, due to the relatively simple microbial community and extreme characteristic, studies of microbial diversity in AMDs have increased in theoretical research, commercial and environmental applications of AMD-derived microorganisms during the past several decades [8-11].
Previous 16S rDNA gene-based phylogenetic studies showed that microbial communities in AMDs consisted of a mixture of various microorganisms, whose community structures were mainly shaped by surrounding geochemical variables, such as pH, S, and metal ions, (Pb, Zn, Cu and Fe ions) [1, 12]. However, few studies performed a comprehensive multivariate analysis for the influence of geochemical variables on the bacterial communities in AMD. In addition, geochemical variables and spatial distance were thought to be the other important factors affecting microbial ecology in soils [13-16]. However, it is still unknown how spatial distance affects microbial communities in AMDs.
The objectives of this study are: 1) Understanding the bacterial community diversity and structure in AMD obtained from three Chinese copper mines; 2) Accessing how geochemical variables and spatial distance shape such AMD microbial community structure. A detailed description of microbial diversity and important implications for understanding the influence of geochemical variables and spatial distance on bacterial communities are provided.
2 Materials and methods
2.1 Site description and sampling
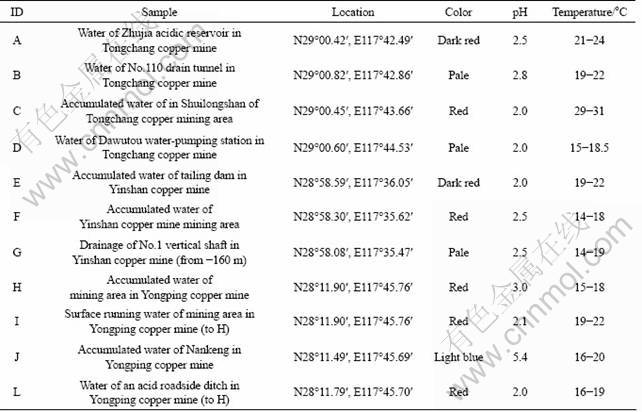
A total of eleven AMD samples were collected from three copper mines, that is, Tongchang Mine (TC), Yinshan Mine (YS) and Yongping Mine (YP) [17], located in southeast of China at April, 2007 (Table 1). Samples A-D were collected from TC. Sample A was obtained from the Zhujia acidic reservoir, the 0.05-0.10 grade Cu ores were piled up near this reservoir and pure Cu was routinely recovered from cycled AMD in this reservoir using biohydrometallurgical technologies since 1998. It produced 1 500 t and 1 400 t pure Cu in 2005 and 2006, repectively [18]. Sample B was mixture of rain and accumulated AMD collected from the No.110 drain tunnel, a part of the sewage treatment system in TC. Sample C was collected from accumulated AMD pond near the open pit. Sample D was collected from Dawutou water-pumping station. Samples E, F, and G were collected from the tailing dam, accumulated water pool in the open pit, and drainage of No.1 vertical shaft (-160 m) of YS, respectively. Samples H, I, J, L were collected from accumulated water pool of mining area, surface running water near sample H, accumulated water of Nankeng, and an acid roadside ditch of YP, respectively.
Approximately, 50 L of water was collected from three position (as three replicates) of each site and transported to laboratary within 48 h at room temperature. Due to the low biomass and DNA yeild, we combined the three replicate into one. A total of 50 mL of the original water sample was used for geochemical analysis. All water samples were then filtered through a sterile 0.22 ��m hyperfiltration membrane. These filters were then immediately transferred to a tube and stored at -20 ��C until they were used for analysis.
Latitude and longitude of sampling sites were determined using a hand holding GPS, Garmin eTrex (Garmin, Kansas, USA). The temperature and pH of each sampling site were measured on sites. A total of 20 elements were measured using inductively coupled plasma-atomic emission spectrometry (ICP-AES; PS-6 Plasma Spectrovac, Baird, USA).
2.2 DNA extraction and purification
Total community DNA was extracted from filtered sediment using a protocol described by ZHOU et al [19]. The crude DNA was further purified using the Promega Wizard DNA Clean-Up System (Madison, WI, USA) according to the manufacturer��s instruction. DNA quality was evaluated by the absorbance ratios at A260/280 and A260/230 using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies Inc., Wilmington, DE). The PicoGreen method [20] was used for quantifying DNA concentration using a FLUOstar Optima (BMG Labtech, Jena, Germany). Purified DNA was stored at -80 ��C until it was used.
Table 1 Sampling description, location and physical properties of studied AMD samples in three mine areas in China
2.3 PCR amplification, purification, 16S rDNA gene cloning and sequencing
The bacterial 16S rDNA genes were amplified and PCR products were purified using the QIAquick Gel Extraction Kit (QIAGEN, Valencia, CA), ligated into pCR2.1 vector (Invitrogen Corporation, Carlsbad, CA), and then transformed into One Shot TOP10 Chemically Competent (Invitrogen Corporation, Carlsbad, CA). Plasmid clones were identified based on blue-white screening and they were grown overnight on plates with ampicillin (100 mg/mL) and X-gal (15 mg/mL). White colonies from each of these libraries were randomly selected and the 16S rDNA gene sequences were determined at the Genome Sequencing Center of Washington University (St. Louis, Missouri, USA).
2.4 Phylogenetic analysis
The nucleotide sequences were assembled and edited using Sequencer v.4.8 (GeneCodes, Ann Arbor, MI) and manually checked for chimeras using Ribosomal Database Project II. Identified chimeric sequences were discarded. Operational taxonomic units (OTUs) were determined using DOTUR [21] with a 3% cutoff value. Phylogenetic trees were generated by using neighbor- joining analysis which show the phylogenetic relationships of bacterial 16S rRNA gene sequences recovered from these AMD samples. The fraction number after the GenBank accession number represents the number of clones for each phylotype versus the total number of clones analyzed in each sample. Scale bars indicate the Jukes-Cantor distances. Bootstrap values of >50% (for 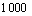 iterations) are shown. Aquifex pyrophilius is used as outgroup. The coverage of clone libraries was calculated using the equation: Rcov=1-(n/N), where n is the number of clones that occurred only once and N is the total number of analyzed clones in each clone library [22].
iterations) are shown. Aquifex pyrophilius is used as outgroup. The coverage of clone libraries was calculated using the equation: Rcov=1-(n/N), where n is the number of clones that occurred only once and N is the total number of analyzed clones in each clone library [22].
2.5 Statistical methods
Principal component analysis (PCA) and detrended correspondence analysis (DCA) were performed to analyze the geochemical variables and community structure with CANOCO (Version 4.5, Biometris-Plant Research International, Netherlands), respectively [23]. Both Shannon-Weaver H and Simpson were calculated using R 2.9.1 (http://www.r-project.org/) to evaluate the community diversity. Shannon�CWeaver index was calculated used the formula: H=-��(pi)(log2pi) and Simpson��s index was calculated based on the formula: 1-D=1- where pi is the proportional abundance of the clone sequenced in each samples. The geographic paired distances of all samples were calculated with the latitude and longitude data using web-based program (http://www.movable-type.co.uk/scripts/latlong. html). Geochemical variables of AMDs were z transformed to convert all measurements to the same scale prior to further analysis using the formula,
where pi is the proportional abundance of the clone sequenced in each samples. The geographic paired distances of all samples were calculated with the latitude and longitude data using web-based program (http://www.movable-type.co.uk/scripts/latlong. html). Geochemical variables of AMDs were z transformed to convert all measurements to the same scale prior to further analysis using the formula,  where xi is the sample value,
where xi is the sample value, 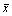 is the mean of all samples, and s is the standard deviation [24]. Mantel and partial Mantel tests [25] for linking phylogenetic microbial community, geochemistry and geographic distance were performed with the vegan package in R 2.9.1. Canonical correspondence analysis (CCA) [26] was also performed with CANOCO. Stepwise CCA was performed to eliminate the redundant geochemical variables based on their inflation factors [24]. CCA was performed with focus on interspecies distance and significance was tested using the Monte Carlo permutation (499 permutations). The partial CCA (pCCA) [23] was used to partition variation in community composition to geochemical variables and geographic distances.
is the mean of all samples, and s is the standard deviation [24]. Mantel and partial Mantel tests [25] for linking phylogenetic microbial community, geochemistry and geographic distance were performed with the vegan package in R 2.9.1. Canonical correspondence analysis (CCA) [26] was also performed with CANOCO. Stepwise CCA was performed to eliminate the redundant geochemical variables based on their inflation factors [24]. CCA was performed with focus on interspecies distance and significance was tested using the Monte Carlo permutation (499 permutations). The partial CCA (pCCA) [23] was used to partition variation in community composition to geochemical variables and geographic distances.
All of the nucleotide sequences obtained in this study had been deposited to the GenBank database under accession numbers: FJ825151-FJ825286 and FJ869157- FJ869171.
3 Results
3.1 Geochemical characteristics of samples
The temperature of the eleven sites ranged from 16 ��C to 30 ��C. Sample C had the highest temperature (30 ��C) with almost 10 ��C higher than other AMDs, and pH was not substantially different within a range of 2-3 except for sample J (pH 5.4) (see Table 1). A total of 20 chemical elements were analyzed by the ICP-AES in the studied eleven AMD samples (see Table 2). These eleven samples are heterogeneous in terms of geochemical components but all are rich in sulfur (0.92-14.03 g/L), and metals such as Fe (0.001-9.890 g/L), Al (0.002- 2.850 g/L ), Cu (0.002-0.490 g/L ), Mg (0.10-2.42 g/L), Mn (0.012-0.660 g/L), Na (0.005-0.110 g/L) and Ca (0.11-0.59 g/L). Samples E, F and G have much higher concentrations of Zn (0.10-0.57 g/L) than other AMDs, and this may be due to the fact that they are close to each other at the same copper mine. In general, sample E has the highest concentration and sample J has the lowest concentration for most elements, especially S, Fe, Al, Mn, and Cu (see Table 2), and all of these eleven AMD samples can be divided into two groups (RLMR and RHMR AMD) according to the total amount of metal content. These also indicate PCA with 77.6% of the total variance explained by the first axis (PC1) and the second axis (PC2) (see Fig.1). Interestingly, two groups are clustered together due to their total metal concentrations, one RHMR group including sample A, C, E, H, I and L, and the other RLMR group including samples B, D, F, G and J (see Fig.1).
Table 2 Geochemical element properties of collected AMD samples (mg/L)

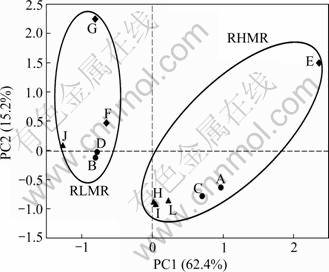
Fig.1 PCA of geochemical variables
3.2 Overall phylogenetic microbial community
There are a total of 1 691 clone sequences obtained from the eleven AMD samples. In order to test whether the analyzed clones can represent the bacteria community in each sample, the coverage of all clone libraries (see Table 3) was calculated. The results show that the obtained data can represent the real status of bacterial communities in those habitats. According to the phylogenetic analysis of 16S rDNA, all 16S rDNA sequences fall into ten known clusters (��-Proteobacteria, ��-Proteobacteria, ��-Proteobacteria, Acidobacteria, Actinobacteria, Bacteriodetes, Cyanobacteria, Firmicutes, Nitrospirae, and Planctomycetes) and some unknown bacterial clusters (see Fig.2). The lowest numbers of clone sequences are obtained from sample I, which has been obtained from the running AMD in mine area. The highest numbers of clone sequences are obtained from sample G, which has been obtained from -160 m level of vertical shaft (see Table 3). The phylogenetic diversity was evaluated for each sample from the clone data by Shannon Weaver ��H�� and evenness. The results show that sample A has the highest diversity, while the diversity in sample L has the lowest, less than three time that of sample A (see Table 3). These results suggest that microbial communities in different AMDs are heterogeneous.
Table 3 Total detected number and coverage of clone sequences and diversity indices of bacterial 16S rRNA gene clones retrieved from studied AMD samples


Fig.2 Relative abundance of all detected OTU
DCA of OTUs were also used to examine overall microbial community structures (see Fig.3). The total inertia for the model is 1.607. Eigenvalues for the first two axes of the DCA are 0.484 and 0.212, respectively, and explain the 43.2% of the variance. Interestingly, the DCA pattern of microbial community structure is roughly clustered into two main groups which are consistent with the PCA pattern of geochemical variables. For the RHMR AMD samples (A, C, E, H, I and L), ��-Proteobacteria, Acidobacteria, Actinobacteria, Cyanobacteria, Firmicutes and Nitrospirae are dominant. In the RLMR AMD samples (B, D, F, G and J), ��- Proteobacteria, ��-Proteobacteria, Planctomycetes and Bacteriodetes are dominant.
3.3 Relative abundance of phylogenetic groups
The relative abundance of the clones related to each major group were calculated based on the number of clone sequences, and the results show that the microbial community structure is in high diversity and heterogeneous in eleven AMDs (Figs.2 and 3). For RLMR AMD samples, the major (relative abundance >30%) groups are ��-Proteobacteria (50.8%) for sample B; Actinobacteria (42.5%) for sample D; Firmicutes (36.7%) for sample F; ��-Proteobacteria (68.6%) for sample G and ��-Proteobacteria (80.4%) for sample J. For RHMR AMD samples, the major groups are Firmicutes (37.7%) for sample A; Actinobacteria (43.5%) for sample C; ��-Proteobacteria (62.9%) for sample E; ��-Proteobacteria (30.6%), Actinobacteria (43.3%) for sample H; ��-Proteobacteria (35.6%) and Actinobacteria (30%) for sample I and Actinobacteria (87.2%) for sample L (Fig.2 and Table 4).

Fig.3 DCA result of detected OTU
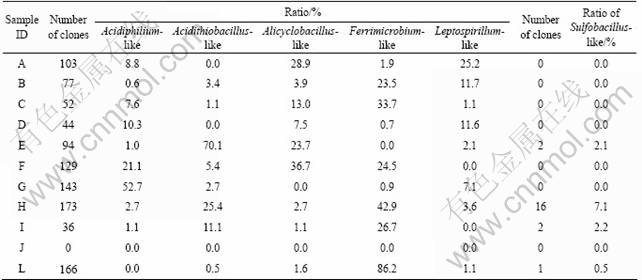
About 60.1% (1 017 out of 1 691) of all clone sequences retrieved in this work are affiliated with putative iron-oxidizing strains or iron-oxidizing bacteria- related clones, falling into the Acidiphilium, Acidithiobacillus, Alicyclobacillus, Ferrimicrobium, and Leptospirillum genera (see Table 5). More than 50% of the clones from the samples A, C, E, F, G, H, and L are affiliated into putative iron-oxidizing bacteria and most of these samples are from RHMR AMDs. However, no putative iron-oxidizing bacteria are found in the sample J, in which pH is 5.4. In contrast, only a small proportion (about 1%, 21 out of 1 691) of retrieved clones are related to putative sulfur-oxidizer-originated sequences and are only present in the samples E, H, I and L, all of them from RHMR AMDs. The results indicate that putative iron-oxidizing may be dominant in high heavy metal enriched AMDs and the few putative sulfur- oxidizing microorganisms are only in rich heavy metal samples. All of these results suggest that iron oxidizing bacteria might contribute a lot for AMD generation since 60% secquences of them are detected.
3.4 Linkages between phylogenetic microbial community and geochemical variables, spatial distribution
Partial Mantel tests were performed to analyze the relationships between AMD microbial community and their spatial distance or surrounding geochemistry. The results show that geochemical variables (r=0.429 3, P= 0.037 7) and spatial distance (r=0.321 3, P=0.018 1) are significantly positively correlated with microbial community. The simple Mantel test between geochemical variables and spatial distance shows that they are independent variables and not correlated (r=-0.04 0, P= 0.565) with each other. These results indicate that surrounding environment and spatial distribution are two independent variables and both of them are associated with the AMD microbial communities.
Table 4 Relative abundance (%) of clones affiliated with each major group
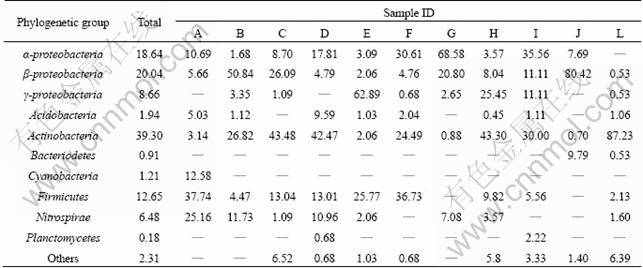
Table 5 Bacterial clones affiliated with putative iron-oxidizing or sulfur-oxidizing bacteria in each clone library
To determine the key geochemical variables shaping microbial community structure, CCA was performed with the numbers of OTUs and geochemical variables. The stepwise selection based on the variance inflation factors (VIFs) for geochemical variables was performed to eliminate the high variance inflation factor (VIF) geochemical variables [24] and finally, pH, Na, Mg, Fe, K and Ca were selected for CCA. The specified CCA model extracting the maximum relationship between community composition and the geochemical variables explains 48.4% of the variation (sum of all eigenvalues, 4.679) and is significant (p=0.01) (see Fig.4).

Fig.4 CCA numbers of all sequenced OTU (symbols) and environmental variables (arrows) (p=0.01)
Variation partitioning analysis (VPA) [13, 27] was performed to better understand how geochemical variables and spatial distance influence the microbial community. The same geochemical variables and pair-wise distance data used for CCA were used for VPA. The results show that geochemical variables are able to independently explain 8% of the variation observed while spatial distance explains 22%. Interactions between these two variables have more influence in this system than individual variables (62%). About 8% of the variation cannot be explained. These results suggest that both geochemistry and spatial distance greatly influence the microbial community.
4 Discussion
AMD is an extremely aquatic environment with low pH and high heavy metal concentration on Earth [1]. It is important to investigate this extreme environment because it may help us not only better understand the life forms in extreme environments but also develop strategies for AMD bioremediation. Also, it may provide useful information in bioleaching, which is a technique using the low pH and high heavy metal tolerated microorganisms to extract metals from ores to solution [1, 28]. In this work, AMDs from three copper mines in southeast China were obtained and the compositions of microbial communities were investigated. Moreover, multivariate analysis was performed to analyze the relationship among the geochemical variables, spatial distribution and microbial communities.
The geochemical property of AMDs is heterogeneous and contains lots of metals. Since S, Cu, Fe, Zn and Pb are the major elements hosted by sulfides, most of the AMD contains those heavy metals [29-30]. The sulfides in most coal and metal deposits also contain trace elements, such as Mn, Cr, and Co, whose concentrations will increase as the dissolution of sulfides proceeds [29-30]. In addition, AMD may also react with other minerals, such as silicates, which will lead to an increase in the dissolved loading of Na, K, Mg, Ca, Si, and Al [31]. In the studied AMDs, there are very high concentrations of S, Fe, Al, Mn, Mg, Cu, Zn, Ca, and lots of trace elements, which is consistent with the previous reports [1, 28, 32]. AMDs are different with respect to their geochemical backgrounds (i.e. element composition in bedrock, and wall rock) which depend upon the location of the mining site, thus the element concentration of the AMDs varies among different locations all over the world [31, 33]. In this work, the consistent results are obtained, for example, although samples C and D are from the same copper mine area and acidic (2.0), the elemental composition is different. Moreover, according to the PCA of geochemical variables, samples from the same mine area cannot be clustered together. However, total element concentration of the geochemical variables and PCA results show that all samples can be clustered into two groups according to their concentrations, one is high metal rich AMD sample group (including samples A, C, E, H, I and L) and the other is low metal rich AMD sample group (including samples B, D, F, G and J). The pH of studied AMDs is low, which is around 2-3 except sample J (pH=5.4). In all studied AMDs, the temperature is not high, because the sampling time is April, 2007; it is still cold in mining area, except the sample C which reaches 30 ��C.
It is expected that the major components of the bacterial community in the investigated AMD samples are consistent with previous studies in other Chinese metal mine AMDs, in which clone sequences are affiliated with ��-Proteobacteria, ��-Proteobacteria, and ��-Proteobacteria, Acidobacteria, Actinobacteria, Firmicutes and Nitrospirae [28, 34]. However, the relative abundance of these major groups varies with different AMDs although the AMD samples are of similar pH and/or temperature. For example, in Ref.[28], the YSK3 (pH 2.5; temperature 20 ��C) sample (collected from Yinshan Mine) contained two major groups (��-Proteobacteria and Nitrospirae, corresponding to 53.1% and 41.3%, respectively). In contrast, the sample E and sample F (pH 2.5; temperature 16.5 ��C) in this work are also taken from Yinshan Mine, but contain different major groups of microorganisms: The sample E comprises of ��-Proteobacteria (62.89%) and Firmicutes (25.77%); the sample F is composed of ��-Proteobacteria (30.61%), Actinobacteria (24.49%) and Firmicutes (36.73%). This difference may be caused by the different sampling sites in mining area and different sampling season. For studied AMDs, we sampled in April, 2007, and for YSK samples, they were collected in August, 2006. It is reported that seasonal changes in pH have significant effects on the aspects of the microbial community in AMD [35].
The extreme condition (e.g. low pH, and high-level metal ions) of AMD results primarily from the metabolic activities of chemolithotrophic microorganisms [36], such as iron-oxidizing prokaryotes (e.g. Acidithiobacillus ferrooxidans, Leptospirillum spp., Ferroplasma acidiphilum), sulfur-oxidizing prokaryotes (e.g. Acidithiobacillus thiooxidans, Sulfolobus shibitae), and some heterotrophic prokaryotes (e.g. Acidiphilium spp., Alicyclobacillus spp.) [32-33]. In this work, about 60.1% of all clone sequences retrieved are affiliated with putative iron-oxidizing strains or iron-oxidizing bacteria-related clones, and only a small proportion (about 1%) of retrieved clones are related to putative sulfur-oxidizer-originated sequences. Interestingly, most of putative iron-oxidizing strains or iron-oxidizing bacteria-related clones and putative sulfur-oxidizer- originated clones sequences are detected from high-level heavy metal enriched AMDs. This evidence suggests that putative iron-oxidizing and sulfur-oxidizing strains contribute a lot for the generation of AMD. Controlling or enhancing the growth of these bacteria might be important to bioremediation for AMD or bioleaching, respectively.
DCA of the microbial communities indicates that microbial communities can be separated into two groups and different dominant species are observed in high and low heavy metal rich AMDs, whose patterns are marginally similar with PCA of the geochemical variables. These results suggest that there are some linkage between the microbial community and geochemical variables. Mantel test and CCA validate this point, which shows that geochemical variables have a significant effect on microbial communities. CCA also shows that the key geochemical variables (pH, Na, Mg, Fe, K and Ca) significantly affect the microbial communities. The previous research has shown that microbial community structure within AMD is influenced by surrounding geochemical variables, such as, pH, S, Pb, Zn, Cu, Ca and ferrous iron [1, 34]. The results are consistent with the previous study; furthermore, Na, Mg and K are also significantly correlated with microbial community. The previous studies showed that Ca, Co, Na, Fe, Mg, and K are essential elements and Mn, Cr, Cu and Zn are required in trace amounts for cell growth [37]. However, there were few literatures that reported the influence of surrouding environmental factors shaping AMD microbial community and the mechnism of it is still unclear. In addition to geochemical variables, spatial distance is also significantly correlated with microbial community. Spatial distance is thought to be another important factor affecting microbial ecology in soils [13-16]. However, few studies are performed to investigate the influence of spatial distance for AMD microbial communities. Variation partitioning analysis (VPA) [27] results show that 92% of the variation could be explained by geochemical variables and spatial distance, and only 8% of the variation cannot be explained. These results show that all the geochemical variables and spatial distance can explain most of the variations in microbial community, and the unexplained part may be due to the distribution of nutrients, carbon sources, ions, or terminal electron acceptors in AMD which are not detected.
5 Conclusions
1) AMDs in this work are heterogeneous and enriched with S, Fe, Al, Mg, Ca, Cu, Mn, Zn and other trace elements. Moreover, these AMDs can be classified into relatively high and low metal rich AMDs according to the total heavy metal concentration and PCA.
2) The microbial communities in AMDs are heterogeneous and all 16S rDNA sequences fall into ten clusters (��-Proteobacteria, ��-Proteobacteria, ��-Proteobacteria, Acidobacteria, Actinobacteria, Bacteriodetes, Cyanobacteria, Firmicutes, Nitrospirae, and Planctomycetes) and some unknown bacterial clusters according to the phylogenetic analysis of 16S rDNA genes.
3) Microbial communities in high metal rich AMDs vary to low metal rich AMDs according to the DCA. For the RHMR AMD samples (A, C, E, H, I and L), ��-Proteobacteria, Acidobacteria, Actinobacteria, Cyanobacteria, Firmicutes and Nitrospirae are dominant. In the RLMR AMD samples (B, D, F, G and J), ��- Proteobacteria, ��-Proteobacteria, Planctomycetes and Bacteriodetes are dominant. The putative iron-oxidizing is dominant and a few putative sulfur-oxidizing microorganisms are only found in RHMR AMDs.
4) Both geochemical variables (r=0.429 3, P=0.037 7) and spatial distance (r=0.321 3, P=0.018 1) have a positive correlation with microbial composition of AMDs. The geochemical variables, such as, pH, Na, Mg, Fe, K and Ca are most important in shaping the microbial community in AMDs. Variation partitioning analysis shows that geochemical variables and spatial distance could explain 92% of total variation in microbial community in AMD.
References
[1] BAKER B J, BANFIELD J F. Microbial communities in acid mine drainage [J]. Fems Microbiol Ecol, 2003, 44(2): 139-152.
[2] GERHARDT A, DE BISTHOVEN L J, GUHR K, SOARES A M V M, PEREIRA M J. Phytoassessment of acid mine drainage: Lemna gibba bioassay and diatom community structure [J]. Ecotoxicology, 2008, 17(1): 47-58.
[3] TREMAINE S C, MILLS A L. Impact of water column acidification on protozoan bacterivory at the lake sediment-water interface [J]. Appl Environ Microb, 1991, 57(3): 775-784.
[4] DSA J V, JOHNSON K S, LOPEZ D, KANUCKEL C, TUMLINSON J. Residual toxicity of acid mine drainage- contaminated sediment to stream macroinvertebrates: Relative contribution of acidity vs. metals [J]. Water Air Soil Poll, 2008, 194(1/2/3/4): 185-197.
[5] LEVINGS C D, BARRY K L, GROUT J A, PIERCEY G E, MARSDEN A D, COOMBS A P, MOSSOP B. Effects of acid mine drainage on the estuarine food web, Britannia Beach, Howe Sound, British Columbia, Canada [J]. Hydrobiologia, 2004, 525(1/2/3): 185-202.
[6] NORDSTROM D K, ALPERS C N. Negative pH, efflorescent mineralogy, and consequences for environmental restoration at the Iron Mountain Superfund site, California [J]. Proc Natl Acad Sci USA, 1999, 96(7): 3455-3462.
[7] TIWARY R K. Environmental impact of coal mining on water regime and its management [J]. Water Air Soil Poll, 2001, 132(1/2): 185-199.
[8] JOHNSON D B, HALLBERG K B. Acid mine drainage remediation options: A review [J]. Sci Total Environ, 2005, 338(1/2): 3-14.
[9] VALENZUELA L, CHI A, BEARD S, ORELL A, GUILIANI N, SHABANOWITZ J, HUNT D F, JEREZ C A. Genomics, metagenomics and proteomics in biomining microorganisms [J]. Biotechnol Adv, 2006, 24(2): 197-211.
[10] CID C, GARCIA-DESCALZO L, CASADO-LAFUENTE V, AMILS R, AGUILERA A. Proteomic analysis of the response of an acidophilic strain of Chlamydomonas sp (Chlorophyta) to natural metal-rich water [J]. Proteomics, 2010, 10(10): 2026-2036.
[11] KNOB A, CARMONA E C. Purification and characterization of two extracellular xylanases from penicillium sclerotiorum: A novel acidophilic xylanase [J]. Appl Biochem Biotech, 2010, 162(2): 429-443.
[12] WAN Ming-xi, YANG Yu, QIU Guan-zhou, XU Ai-ling, QIAN Lin, HUANG Zhi-ying, XIA Jin-lan. Acidophilic bacterial community reflecting pollution level of sulphide mine impacted by acid mine drainage [J]. Journal of Central South University of Technology, 2009, 16(2): 223-229.
[13] RAMETTE A, TIEDJE J M. Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem [J]. Proc Natl Acad Sci USA, 2007, 104(8): 2761-2766.
[14] HORNER-DEVINE M C, LAGE M, HUGHES J B, BOHANNAN B J M. A taxa-area relationship for bacteria [J]. Nature, 2004, 432(7018): 750-753.
[15] MARTINY J B H, BOHANNAN B J M, BROWN J H, COLWELL R K, FUHRMAN J A, GREEN J L, HORNER-DEVINE M C, KANE M, KRUMINS J A, KUSKE C R, MORIN P J, NAEEM S, OVREAS L, REYSENBACH A L, SMITH V H, STALEY J T. Microbial biogeography: Putting microorganisms on the map [J]. Nat Rev Microbiol, 2006, 4(2): 102-112.
[16] ZHOU Ji-zhong, KANG S, SCHADT C W, GARTEN C T. Spatial scaling of functional gene diversity across various microbial taxa [J]. Proc Natl Acad Sci USA, 2008, 105(22): 7768-7773.
[17] XIE Jian-ping, HE Zhi-li, LIU Xin-xing, VAN NOSTRAND J D, DENG Ye, WU Li-you, ZHOU Ji-zhong, QIU Guan-zhou. GeoChip-based analysis of the functional gene diversity and metabolic potential of microbial communities in acid mine drainage [J]. Appl Environ Microbiol, 2011, 11(3): 991-999.
[18] QIU Guan-zhou, WANG Jun. Bio-leaching of low-grade large porphyry chalcopyrite-containing ore [J]. Transactions of Nonferrous Metals Society of China, 2000, 10(s1): 19-22.
[19] ZHOU Ji-zhong, BRUNS M A, TIEDJE J M. DNA recovery from soils of diverse composition [J]. Appl Environ Microb, 1996, 62(2): 316-322.
[20] AHN S J, COSTA J, EMANUEL J R. PicoGreen quantitation of DNA: Effective evaluation of samples pre- or post-PCR [J]. Nucleic Acids Res, 1996, 24(13): 2623-2625.
[21] SCHLOSS P D, HANDELSMAN J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness [J]. Appl Environ Microb, 2005, 71(3): 1501-1506.
[22] HUMAYOUN S B, BANO N, HOLLIBAUGH J T. Depth distribution of microbial diversity in Mona Lake, A mermictic soda lake in California [J]. Appl Environ Microb, 2003, 69(2): 1030-1042.
[23] TERBRAAK C J F. Canoco��An extension of decorana to analyze species-environment relationships [J]. Vegetatio, 1988, 75(3): 159-160.
[24] van NOSTRAND J D, WU Wei-min, WU Li-you, DENG Ye, CARLEY J, CARROLL S, HE Zhi-li, GU Bao-hua, LUO Jian, CRIDDLE C S, WATSON D B, JARDINE P M, MARSH T L, TIEDJE J M, HAZEN T C, ZHOU Ji-zhong. GeoChip-based analysis of functional microbial communities during the reoxidation of a bioreduced uranium-contaminated aquifer [J]. Environ Microbiol, 2009, 11(10): 2611-2626.
[25] MANTEL N. Detection of disease clustering and a generalized regression approach [J]. Cancer Res, 1967, 27(2p1): 209-220.
[26] HOTELLING H. Relations between two sets of variates [J]. Biometrika, 1936, 28(3/4): 321-377.
[27] OKLAND R H, EILERTSEN O. Canonical correspondence-analysis with variation partitioning��Some comments and an application [J]. J Veg Sci, 1994, 5(1): 117-126.
[28] YIN Hua-qun, CAO Lin-hui, XIE Ming, CHEN Qi-jiong, QIU Guan-zhou, ZHOU Ji-zhong, WU Li-you, WANG Dian-zuo, LIU Xue-duan. Bacterial diversity based on 16S rRNA and gyrB genes at Yinshan mine, China [J]. Syst Appl Microbiol, 2008, 31(4): 302-311.
[29] MATLOCK M M, HOWERTON B S, ATWOOD D A. Chemical precipitation of heavy metals from acid mine drainage [J]. Water Res, 2002, 36(19): 4757-4764.
[30] SCHMIDT A, HAFERBURG G, SINERIZ M, MERTEN D, BUCHEL G, KOTHE E. Heavy metal resistance mechanisms in actinobacteria for survival in AMD contaminated soils [J]. Chem Erde-Geochem, 2005, 65(S1): 131-144.
[31] DINELLI E, TATEO F. Different types of fine-grained sediments associated with acid mine drainage in the Libiola Fe-Cu mine area (Ligurian Apennines, Italy) [J]. Appl Geochem, 2002, 17(8): 1081-1092.
[32] ALLEN E E, BANFIELD J F. Community genomics in microbial ecology and evolution [J]. Nat Rev Microbiol, 2005, 3(6): 489-498.
[33] JOHNSON D B. Biodiversity and ecology of acidophilic microorganisms [J]. Fems Microbiol Ecol, 1998, 27(4): 307-317.
[34] YIN Hua-qun, QIU Guan-zhou, WU Li-you, XIE Ming, ZHOU Ji-zhong, DAI Zhi-min, WANG Dian-zuo, KELLOGG L, CAO Lin-hui, LIU Xue-duan. Microbial community diversity and changes associated with a mine drainage gradient at the Dexing copper mine, China [J]. Aquat Microb Ecol, 2008, 51(1): 67-76.
[35] EDWARDS K J, GIHRING T M, BANFIELD J F. Seasonal variations in microbial populations and environmental conditions in an extreme acid mine drainage environment [J]. Appl Environ Microb, 1999, 65(8): 3627-3632.
[36] SINGER P C, STUMM W. Acidic mine drainage: The rate- determining step [J]. Science, 1970, 167(3921): 1121-1127.
[37] FINNEY L A, O'HALLORAN T V. Transition metal speciation in the cell: Insights from the chemistry of metal ion receptors [J]. Science, 2003, 300(5621): 931-936.
(Edited by DENG L��-xiang)
Foundation item: Project(2010CB630901) supported by the National Basic Research Program of China; Project(50621063) supported by Creative Research Group of China; Projects(51104189, 50321402, 50774102) supported by the National Natural Science Foundation of China; Project (1343-77341) supported by the Graduate Education Innovative Program of Central South University, China; Project(DOE-ER64125) supported by the Department of Energy, Office of Science under the Environmental Remediation Science Program of USA
Received date: 2010-10-13; Accepted date: 2010-12-20
Corresponding author: XIE Jian-ping, PhD; Tel: +86-13755128216; E-mail: whitewolf1101@gmail.com

