����������Һ��ϵ�е�������ԭ��Ϊ�ͻ�ѧ��̬
��Դ�ڿ����й���ɫ����ѧ��(Ӣ�İ�)2017���9��
�������ߣ������ ����Ԫ ������ �����
����ҳ�룺2063 - 2072
�ؼ��ʣ��飻��-��������λ-pHͼ����ѧ��̬�����Է�ˮ
Key words��arsenic; ferric-arsenic complexes; ��-pH diagram; chemical species; acidic wastewater
ժ Ҫ��ұ�����Է�ˮ�����ȥ����һ��ʮ�ֽ��ȵĹ�����Ŀǰ��õķ������Ƚ�����������Ϊ����飬Ȼ���������ν��г���ȥ�������������ԭ��Ϊ����ѧ��̬�����о��������ȥ�������ش���ѭ��������������ɼ��ֹ��ȷ���չʵ�����о�������HSC��MINTEQ�����������۷������о������������������һ�������ת�Ʒ�Ӧ������ɢ���ƣ���pH 1.0��������Һ�У����������������λ�dz���(Լ0.9 V)�����⣬���Fe(III)- As(V)-H2SO4-H2Oϵ����Һ������-�ɼ������Լ�������̬����������Fe(III)-As(V)�����Ĵ��ڡ���ˣ����������о���Ԥ�������ѧ���ݣ�����Fe(III)��As(V)��������̬��pH�ķֲ�����ͼ�Ͱ���Fe-As���������͵�λ-pHͼ��
Abstract: Arsenic (As) removal from smelting acidic wastewater is an urgent task. The most common method is oxidation of trivalent As(III) to pentavalent As(V) subsequently precipitated by ferric (Fe(III)) salts. Foundations of redox behavior and chemical species are of great importance for understanding As removal. In this work, cyclic voltammetry (CV) and UV-Vis spectroscopy were used for laboratory observation; meanwhile HSC and MINTEQ software were employed for theoretical analyses. It is found that As(III) oxidation, a multiple electron transfer reaction, is diffusion-controlled. The oxidation over-potential is very high (about 0.9 V) in sulfuric acid solutions (pH 1.0). In addition, Fe(III)-As(V) complexes are evidenced by UV-Vis spectra and chemical species analyses in series of Fe(III)-As(V)-H2SO4-H2O solutions. Therefore, the Fe(III) and As(V) species distribution against pH values are determined and a new ��-pH diagram with inclusion of Fe-As complexes is consequently compiled based on thermodynamic data predicted by other researchers.
Trans. Nonferrous Met. Soc. China 27(2017) 2063-2072
Jin-qin YANG1, Li-yuan CHAI1,2, Qing-zhu LI1,2, Yu-de SHU1,2
1. School of Metallurgy and Environment, Central South University, Changsha 410083, China;
2. National Engineering Research Centre for Control and Treatment of Heavy Metal Pollution, Central South University, Changsha 410083, China
Received 16 September 2016; accepted 20 January 2017
Abstract: Arsenic (As) removal from smelting acidic wastewater is an urgent task. The most common method is oxidation of trivalent As(III) to pentavalent As(V) subsequently precipitated by ferric (Fe(III)) salts. Foundations of redox behavior and chemical species are of great importance for understanding As removal. In this work, cyclic voltammetry (CV) and UV-Vis spectroscopy were used for laboratory observation; meanwhile HSC and MINTEQ software were employed for theoretical analyses. It is found that As(III) oxidation, a multiple electron transfer reaction, is diffusion-controlled. The oxidation over-potential is very high (about 0.9 V) in sulfuric acid solutions (pH 1.0). In addition, Fe(III)-As(V) complexes are evidenced by UV-Vis spectra and chemical species analyses in series of Fe(III)-As(V)-H2SO4-H2O solutions. Therefore, the Fe(III) and As(V) species distribution against pH values are determined and a new ��-pH diagram with inclusion of Fe-As complexes is consequently compiled based on thermodynamic data predicted by other researchers.
Key words: arsenic; ferric-arsenic complexes; ��-pH diagram; chemical species; acidic wastewater
1 Introduction
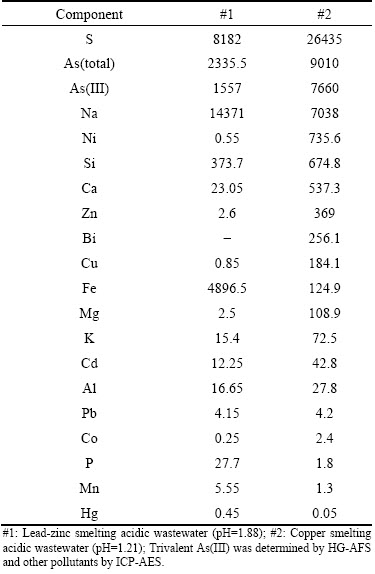
Arsenic (As) is often found in association with nonferrous metal ore [1]. In nonferrous metal smelters, As-containing acidic wastewater was generated from the wet scrubber process of smelting fume [2]. The smelting acidic wastewaters sampled from certain copper smelter and lead-zinc smelter (see Table 1) are very different from common wastewaters, i.e., groundwater, and municipal wastewater [3]. The extremely low pH value, abundant As and complicated composition lead to difficulty in As removal.
One of the potential sources of As is the As2O3- bearing flue dust generated by smelting operations [4,5]. Thus, trivalent state As(III) is predominant, e.g., the ratio of As(III)/As(total) is equal to 67% and 85% in lead-zinc and copper smelting acidic wastewaters, respectively (Table 1). A pre-oxidation treatment is significant because As(III) is more mobile and toxic than pentavalent state As(V) [6]. The oxidation of As(III) to As(V) is unlikely to be a problem for the pressure oxidation systems but difficult under atmosphere [7]. Moreover, the on-site oxidation under strong acidic conditions is challenging. Lime neutralization is commonly used for acidic wastewater treatment. However, lime can produce plenty of gypsum and unstable cadmium arsenate [8]. Consequently, the on-site high-effective oxidation is attractive, which can lay foundation for the subsequent ferric arsenate (FeAsO4) precipitation under strong acidic conditions. As coprecipitation with ferric (Fe(III)) iron has been specified by U.S. EPA as the best demonstrated available technology (BDAT) for the removal of As [9]. Scorodite (FeAsO4��2H2O), ferric arsenate and arsenical ferrihydrite are common precipitates formed from As removal in metallurgical industries [10]. Well-crystalline scorodite is more advantageous in lower ferric demand, higher density and greater stability. The production of scorodite is easily conducted under autoclave conditions, i.e., under high temperature and pressure conditions [11].
However, atmospheric scorodite production is of great concern due to lower capital investment [8]. Works done over last decades by DEMOPOULOS and coworkers [5,12,13] achieved the production of scorodite by step-wise lime neutralization at 90 ��C. In addition, scorodite was precipitated from sulfuric acid at pH 2 at temperatures as low as 70 ��C, but the long reaction time of 16 h was needed [13]. This is because amorphous ferric arsenate is the initial precipitate, which was formed almost immediately and then transformed into scorodite [14]. Previously, it was found that aqueous Fe(III)-As(V) complexes can convert into colloid ferric arsenate under higher temperature and pH conditions [15]. The conversion of aqueous complexes to amorphous ferric arsenate and subsequently to scorodite is meaningful for the removal of As(V) from acidic effluents by coprecipitation with Fe(III). Therefore, thermodynamic chemistry is needed to be studied to provide an insight into the species distribution and transformation in acidic Fe-As system.
In this work, the redox of As in acidic solutions was studied by the combination of cyclic voltammetry (CV) method and theoretical ��-pH diagram analysis. In addition, the species of As in As-H2O and As-Fe-H2SO4-H2O systems were discussed. The new ��-pH diagram for As-Fe-H2SO4-H2O system was constructed with the inclusion of aqueous Fe(III)/Fe(II)- As(V) complexes.
Table 1 Compositions of acidic wastewaters from different smelters (mg/L)
2 Calculation and experiment methods
2.1 Calculation methods
The ��-pH diagram was constructed based on the well-known Nernst equation. A half-cell reaction can be written as follows:
cA+dD+ne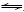 xX+yY (1)
xX+yY (1)
The corresponding Nernst equation was written as Eq. (2). Reversible potential (��0) can be derived from the Gibbs free energy of reaction as Eq. (3):
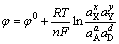 (2)
(2)
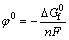 (3)
(3)
where a represents the activity of substances; F is the Faraday��s constant. Software HSC 7.1 was used in this work for the construction of ��-pH diagrams. If not specified, the Gibbs free energies in database of HSC 7.1 were adopted.
Visual MINTEQ 3.1 was used to determinate the speciation-pH diagram. The calculation processes were illustrated by taking As(V) 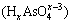 as an example. The formation reaction and corresponding constant equation was shown as Eqs. (4)-(6), where square brackets represent concentration. The sum of
as an example. The formation reaction and corresponding constant equation was shown as Eqs. (4)-(6), where square brackets represent concentration. The sum of  is equal to the total concentration of As(V) (Eq. (7)). The fractions of As(V) species at various pH values were then solved by MINTEQ 3.1.
is equal to the total concentration of As(V) (Eq. (7)). The fractions of As(V) species at various pH values were then solved by MINTEQ 3.1.
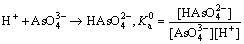 (4)
(4)
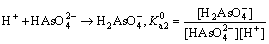 (5)
(5)
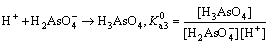 (6)
(6)
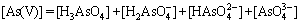 (7)
(7)
Same method was used for the determination of As(III) and Fe(III) species distribution against pH value. If not specified, the formation constants in database of MINTEQ 3.1 were adopted. However, the species distribution against pH can be affected by ionic strength (IS). The equilibrium constant at a given ionic strength 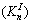 is given by the activity coefficients of the species involved in the equilibrium (Eq. (7)):
is given by the activity coefficients of the species involved in the equilibrium (Eq. (7)):
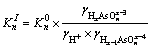 (8)
(8)
In MINTEQ software, IS is calculated based on Eq. (9). The activity coefficient at I<0.3 mol/L is calculated from the Davies equation as Eq. (10):
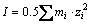 (9)
(9)
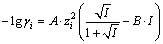 (10)
(10)
where ��i is the activity coefficient of species i, I is the ionic strength, zi is the charge of the species, A is the Debye-H��ckel coefficient, which takes the value of 0.51 at 25 ��C, and B is the so-called Davies B parameter, which has a default value of 0.3.
2.2 Experimental methods
1) Cyclic voltammetry (CV) experiments
For the preparation of experimental solutions, pH 1.0 sulfuric acid was prepared in advance as supporting electrolyte. As(III) was incorporated into solutions by NaAsO2 (Shanghai No. 4 Chemical Reagent Factory, China). Solutions referred to as ��blank�� contained no As. Cyclic voltammetry (CV) experiments were carried out by a three-electrode test system with a gold working electrode, glassy-carbon counter electrode and saturated calomel reference electrode. The solution was placed in a five-necked electrolysis pool in 25 ��C water bath. High purity nitrogen gas was purged into the solutions before applying the potential.
2) UV-Vis spectroscopic experiments
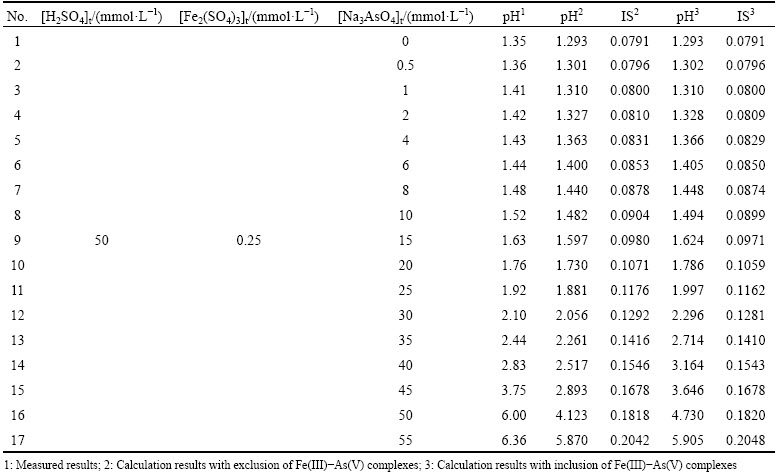
Dilute sulfuric acid solution (50 mmol/L) was prepared using ultrapure water beforehand and used to prepare stock solutions and sample solutions in order to avoid the hydrolysis of Fe(III). The Fe(III) stock solution and As(V) stock solution were prepared by using Fe2(SO4)3 (Xilong, Guangdong Province, China) and Na3AsO4��12H2O (Sinopharm, Shanghai, China), respectively. A series of sample solutions containing 0.5 mmol/L Fe(III) and 0-55 mmol/L As(V) were prepared for UV-Vis tests (Table 2). UV-Vis spectroscopic measurements were performed using a Shimadzu (Japan) UV-2550 double-beam spectro- photometer. A baseline correction was carried out by filling in quartz cells of 1 cm light length with ultrapure water. The spectra were recorded by replacing the ultrapure water with sample solutions in sample cell.
3 Results and discussion
3.1 Redox behavior of As
The ��-pH diagram for As-H2O system was reported in a number of works [16-20], but the stoichiometry of As(III) species was different. HAsO2 stoichiometry was supported by the mass spectrometry, because mass-to-charge ratios (m/z) of 107 corresponding to  ion in the gas phase [21]. However, H3AsO3, a moiety comprising one As atom coordinated by three OH ligands, was supported by Raman and EXAFS spectroscopy [22-24]. Thus, H3AsO3 and its conjugate base were considered here rather than HAsO2. As given in Table 3, the Gibbs free energies of As(III) and As(V) species in HSC 7.0 database are in accordance with those in most reports. Consequently, the ��-pH diagram for As (0.1 mol/L)- H2O system was directly constructed by using HSC 7.0 software.
ion in the gas phase [21]. However, H3AsO3, a moiety comprising one As atom coordinated by three OH ligands, was supported by Raman and EXAFS spectroscopy [22-24]. Thus, H3AsO3 and its conjugate base were considered here rather than HAsO2. As given in Table 3, the Gibbs free energies of As(III) and As(V) species in HSC 7.0 database are in accordance with those in most reports. Consequently, the ��-pH diagram for As (0.1 mol/L)- H2O system was directly constructed by using HSC 7.0 software.
Table 2 Solution composition used in UV-Vis experiment and related calculation results
As depicted in Fig. 1, the predominance fields for As(V) species under oxidizing conditions are similar to all published ��-pH diagrams. The field boundary for As(III) species was different because of the inclusion of  and
and  . Moreover, the critical potentials of As(V)/As(III) couples drop with decreasing pH value, which indicates that the oxidation of As(III) to As(V) is more feasible in alkaline than in acidic solutions. Thus, the redox behavior of As(III) under strong acidic wastewater is challenging. The practice redox behavior of As(III) in sulfuric acid (pH=1.0) was studied by cyclic voltammetry (CV) methods. Figure 2 shows that the voltammogram of a solution contains 0.1 mol/L As(III) and a blank solution. There are six peaks on the CV curve in 0.1 mol/L As(III) solution (blue line). The peaks B and C correspond to the oxygen evolution and the reduction of the oxide layer in comparison with the CV curve in blank solution (red line). The other four peaks A, D, E and F may be related to the redox behavior of As(III). To verify this, a series of CV tests were conducted in As(III)-bearing solutions with different concentrations.
. Moreover, the critical potentials of As(V)/As(III) couples drop with decreasing pH value, which indicates that the oxidation of As(III) to As(V) is more feasible in alkaline than in acidic solutions. Thus, the redox behavior of As(III) under strong acidic wastewater is challenging. The practice redox behavior of As(III) in sulfuric acid (pH=1.0) was studied by cyclic voltammetry (CV) methods. Figure 2 shows that the voltammogram of a solution contains 0.1 mol/L As(III) and a blank solution. There are six peaks on the CV curve in 0.1 mol/L As(III) solution (blue line). The peaks B and C correspond to the oxygen evolution and the reduction of the oxide layer in comparison with the CV curve in blank solution (red line). The other four peaks A, D, E and F may be related to the redox behavior of As(III). To verify this, a series of CV tests were conducted in As(III)-bearing solutions with different concentrations.
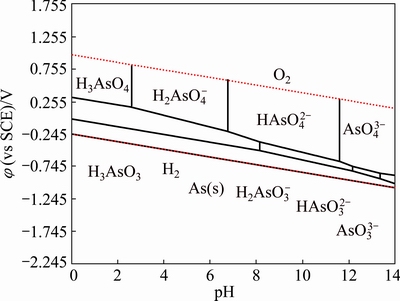
Fig. 1 ��(vs SCE)-pH diagram in As (0.1 mol/L)-H2O system at 25 ��C and 105 Pa
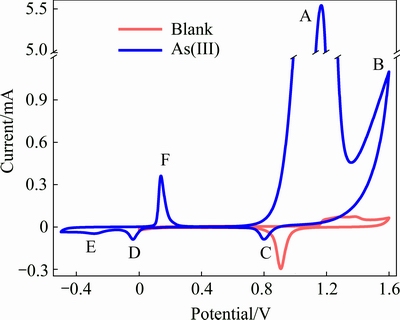
Fig. 2 Voltammogram in 0.1 mol/L As(III) solution (blue line) and blank solution (red line) (��=50 mV/s)
As shown in Fig. 3, the locations of peaks A, D and F all shift to high potential as the concentration of As(III) increases. Moreover, the currents of peak A, D and F increase with As(III) concentration increasing. Peak A obviously splits into three small peaks when As(III) concentration is less than 5 mmol/L. Thus, peak A corresponds to a multiple electron transfer process. Figure 3(b) shows that the current of first electron transfer reaction is linearly correlated with As(III) concentration (R2=0.9985). Peak A is identified as the oxidation of As(III) to As(V). However, the oxidation over-potential of 0.1 mol/L As(III) in sulfuric acid solutions (pH 1.0) is about 0.9 V over the theoretical potential (about 0.3 V derived from Fig. 1). Peak D corresponds to the reduction of As(III), because peak current is linearly correlated to As(III) concentration. However, as shown in Fig. 3(c), the slope of fitting line is different in As(III) ranges of 0-1, 1-2 and 2-5 mmol/L. This indicates that multilayer As(s) is electrodeposited from As(III) on gold electrode. As for the weak peak E, it can be explained as outmost As(s) electrodeposited from As(III) or from As(V). The reduction of As(V) to As(s) is thermodynamic sluggish, but the electrogenerated H2 could chemically reduce As(V) to As(s) [32]. Following peak D and E, the peak F is no doubt the oxidation of As(s) to As(III). Thus, the linear correlation between peak current and As(III) concentration is in accordance with that of peak D. As shown in Fig. 3(c), three lines with different slopes are suitable for fitting in the As(III) concentration ranges of 0-1, 1-2 and 2-5 mmol/L. The consistent trend between peak D and F confirms that As(s) is layeredly electrodeposited on Au electrode. In addition, the oxidation process of As(III) to As(V) was investigated at different scan rates. The good linear relationship between peak current and scan rate (v1/2) (Fig. 3(d)) indicates that As(III) oxidation is mainly diffusion-controlled.
Table 3 Gibbs free energies of 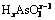 and
and 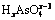 (kJ/mol)
(kJ/mol)
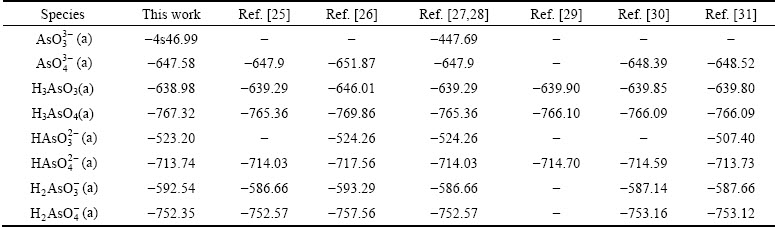
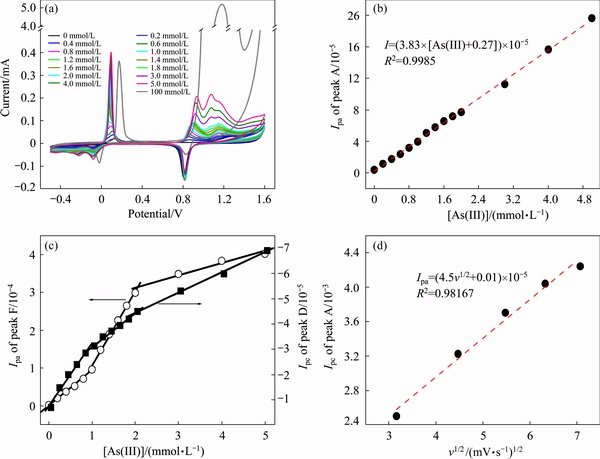
Fig. 3 Voltammograms in As(III) solutions (a), linear calibration plots of peak current against As(III) concentration ([As(III)]=0-5 mmol/L, ��=50 mV/s) (b) and (c), and linear calibration plots of peak A current against v1/2 ([As(III)]=0.1 mol/L, v=10, 20, 30, 40, 50 mV/s) (d)
3.2 Chemical species of As
As(III) and As(V) mainly exist as HxAsO3x-3 and HxAsO4x-3 (0��x��3) in As-H2O system, respectively. The speciation-pH diagrams for As(III) and As(V) were calculated at ionic strength of 0 based on thermodynamic formation constants reported by MARINI and ACCORNERO [33]. The thermodynamic constants 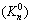 for As(III) and As(V) species are reviewed in Table 4. As given in Table 4, there is no significant difference among different reports. As depicted in Fig. 4(a), As(V) predominantly exists as (1) H3AsO4 and
for As(III) and As(V) species are reviewed in Table 4. As given in Table 4, there is no significant difference among different reports. As depicted in Fig. 4(a), As(V) predominantly exists as (1) H3AsO4 and 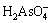 at pH 0-4.5, (2) and
at pH 0-4.5, (2) and 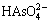 at pH 4.5-9, and (3) and
at pH 4.5-9, and (3) and 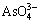 at pH 9-14. The major form of As(V) is neutral molecules H3AsO4 at pH<2, thus it is hard to be removed by electrostatic adsorption. Figure 4(b) shows that As(III) mainly exists as (1) H3AsO3 at pH=0-7.5, (2) H3AsO3 and
at pH 9-14. The major form of As(V) is neutral molecules H3AsO4 at pH<2, thus it is hard to be removed by electrostatic adsorption. Figure 4(b) shows that As(III) mainly exists as (1) H3AsO3 at pH=0-7.5, (2) H3AsO3 and 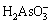 at pH=7.5-10.5, (3) and at pH= 10.5-12 and (4) , and at pH=12-14. Accordingly, H3AsO3 is the only As(III) species under acidic solutions. It is reported that undissociated H3AsO3 does not react with H2O2 [34]. However, rate constant k for the oxidation reaction between As(III) and H2O2 can be determined by pH, temperature and IS at pH=7.5-10.3 [34]. This is mainly because the molar fraction of , and species can be affected by pH, temperature and IS. The speciation-pH diagram at I=0.2 mol/L was determined based on the Davies equation (Eq. (8)). As depicted in Fig. 4, high IS facilitates deprotonation reaction. Therefore, negatively-charged species with higher fraction under high IS solutions can be absorbed on the positive-charged surfaces.
at pH=7.5-10.5, (3) and at pH= 10.5-12 and (4) , and at pH=12-14. Accordingly, H3AsO3 is the only As(III) species under acidic solutions. It is reported that undissociated H3AsO3 does not react with H2O2 [34]. However, rate constant k for the oxidation reaction between As(III) and H2O2 can be determined by pH, temperature and IS at pH=7.5-10.3 [34]. This is mainly because the molar fraction of , and species can be affected by pH, temperature and IS. The speciation-pH diagram at I=0.2 mol/L was determined based on the Davies equation (Eq. (8)). As depicted in Fig. 4, high IS facilitates deprotonation reaction. Therefore, negatively-charged species with higher fraction under high IS solutions can be absorbed on the positive-charged surfaces.
Table 4 Thermodynamic formation constants of and (0��x��3)
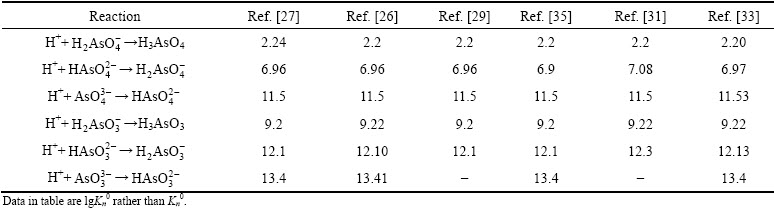
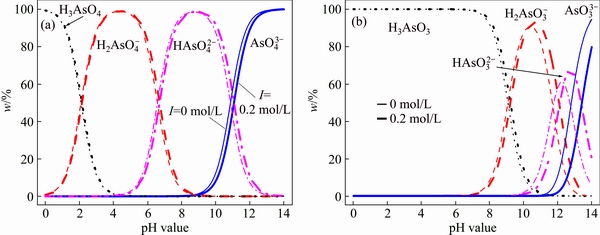
Fig. 4 Speciation-pH diagrams of As(V) species (a) and As(III) species (b) in As-H2O system at 25 ��C
In addition, it is predicted that As(V) can complex with metal ion to form aqueous complexes [33]. Fe(III) is usually used as precipitator, thus the complexation between As(V) and Fe(III) is studied here by UV-Vis spectroscopy. The experimental solutions prepared by Fe2(SO4)3, H2SO4 and Na3AsO4 are all acidic solutions. Figure 5 shows that a new peak at about 240-300 nm appears as As(V) concentration increases. The new peak is attributed to Fe(III)-As(V) complexes. The pH value, IS and the fraction of various Fe(III) species were calculated by MINTEQ 3.1 with the consideration of Fe(III)-As(V) complexes or not. The lgK reported by MARINI and ACCORNERO [36] were adopted here, because they are in accordance with other reports excluding that of FeAsO4(a) (see Table 5). As displayed in Table 2, the calculated pH values are lower than measured ones, but slightly higher when Fe(III)-As(V) complexes are considered.
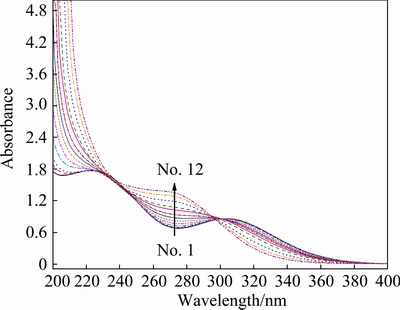
Fig. 5 UV-Vis spectra of Fe2(SO4)3-Na3AsO4-H2SO4-H2O solutions
The Fe(III) species in experimental solutions are shown in Fig. 6. Figure 6(a) shows that Fe(III) predominantly exists as 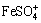 when As(V) concentration is less than 45 mmol/L and as
when As(V) concentration is less than 45 mmol/L and as 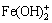 when As(V) concentration is 45-50 mmol/L. It was reported that the band maxima of occur at the wavelength of 300 nm [40]. The new peak at 240- 300 nm is indeed not due to . When Fe(III)- As(V) complexes were considered in the calculations (Fig. 6(b)),
when As(V) concentration is 45-50 mmol/L. It was reported that the band maxima of occur at the wavelength of 300 nm [40]. The new peak at 240- 300 nm is indeed not due to . When Fe(III)- As(V) complexes were considered in the calculations (Fig. 6(b)), 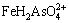 and
and 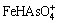 become the major species in solutions with 15-40 mmol/L As(V) and is almost the only soluble Fe(III) species when As(V) concentration is 40-50 mmol/L. In addition, the saturation index is higher than 0.577 when [As(V)]>30 mmol/L. Hematite, goethite, lepidocrocite and FeAsO4��2H2O etc are oversaturated, which can explain that experimental solutions become turbid when As(III) concentration is more than 30 mmol/L. Accordingly, Fe(III)-As(V) complexes are significant to be considered in Fe(III)- As(V)-H2SO4-H2O solutions.
become the major species in solutions with 15-40 mmol/L As(V) and is almost the only soluble Fe(III) species when As(V) concentration is 40-50 mmol/L. In addition, the saturation index is higher than 0.577 when [As(V)]>30 mmol/L. Hematite, goethite, lepidocrocite and FeAsO4��2H2O etc are oversaturated, which can explain that experimental solutions become turbid when As(III) concentration is more than 30 mmol/L. Accordingly, Fe(III)-As(V) complexes are significant to be considered in Fe(III)- As(V)-H2SO4-H2O solutions.
Table 5 Reaction equilibrium constants and Gibbs free energies of Fe-As(V) complexes

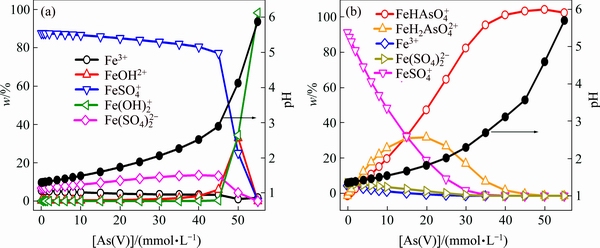
Fig. 6 Fe(III) species analyses in UV-Vis experiment solutions with exclusion (a) and inclusion (b) of Fe(III)-As(V) complexes
As discussed above, Fe(III)-As(V) complexes are important in the species analysis for the system of Fe(III)-As(V)-H2SO4-H2O, a representative system of As(V) removal from acidic wastewater. Then, the speciation-pH diagrams of Fe(III) and As(V) were calculated for such a system with inclusion of Fe(III)-As(V) complexes. As displayed in Fig. 7(a), As(V) mainly exists as (1) and H3AsO4 at pH<1, (2) , , H3AsO4 and at pH=1-2, and (3) at pH>2. As shown in Fig. 7(b), Fe(III) mainly exists as at pH<2, and at pH>2. However, the total dissolved Fe(III) decreases when pH is higher than 2. Therefore, As(V) mainly exists as 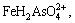 , H3AsO4 and in acidic wastewater treatment system.
, H3AsO4 and in acidic wastewater treatment system.
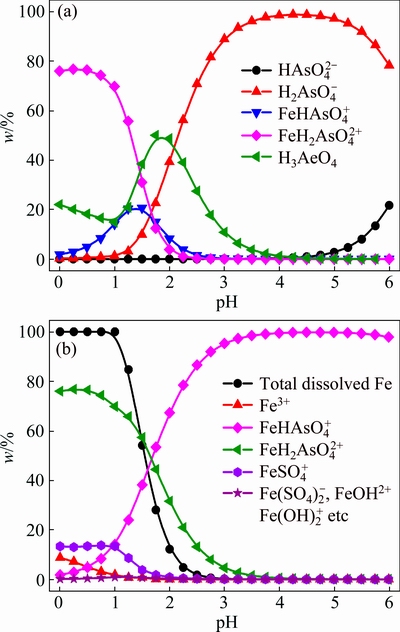
Fig. 7 Speciation-pH diagrams of As(V) (a) and Fe(III) (b) with inclusion of Fe(III)-As(V) complexes ([As(V)]=[Fe(III)]= [SO4]=0.1 mol/L)
3.3 ��-pH diagram in Fe-As-H2O system
As mentioned above, the Fe(III)-As(V) complexes are significant for the speciation of Fe(III) and As(V).
Fe(III)-As(V) complexes should be included in ��-pH diagram for As-Fe-H2O system. According to literatures, Fe(III)-As(III), Fe(II)-As(V) and Fe(II)-As(III) complexes were predicted but there was no experimental evidence. Fe(III)-As(III) and Fe(II)-As(III) complexes are ruled out because of (1) the possible redox reaction between Fe(III) and As(III) and (2) the oxidation of As(III) in parallel to the dark oxidation of Fe(II) by dissolved O2 [41]. Given the complexation ability of As(V), Fe(II)-As(V) complexes were considered here. Therefore, ��-pH diagram for Fe-As-H2O system was constructed with inclusion of Fe(III)-As(V) and Fe(II)-As(V) complexes, and compared with one with exclusion of these complexes.
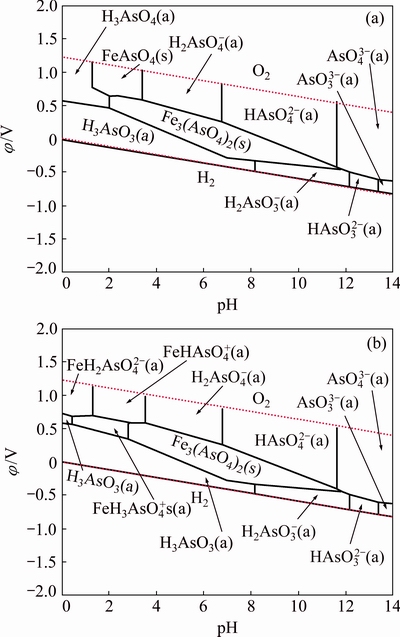
Fig. 8 ��-pH diagrams in 0.1 mol/L As-0.1 mol/L Fe-H2O system with exclusion (a) and inclusion (b) of Fe-As complexes at 25 ��C and 105 Pa (��=��(vs SCE) + 0.245)
The ��-pH diagrams are depicted in Fig. 8 for As-Fe-H2O system at 25 ��C and 105 Pa. Figure 8(a) with exclusion of Fe-As complexes shows that FeAsO4(s) is predominant in the pH range of 1.2-3.4 under oxidizing conditions. It was reported that amorphous ferric arsenate could transform into scorodite [42]. The kinetics of scorodite formation and its transformation from ferric arsenic is strongly controlled by pH, for example, scorodite precipitated after ~384 h at pH 4.5 but ~13 at pH 1 [14]. The pH of smelting acidic wastewater is usually less than 3, which is suitable for the production of ferric arsenate and scorodite. However, Fe(III)-As(V) complexes (,and 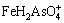 ) were reported predominant under extremely acidic pH condition [43]. Therefore, the ��-pH diagram with inclusion of Fe-As complexes is more meaningful to understand the As geochemistry in Fe-As-H2O system.
) were reported predominant under extremely acidic pH condition [43]. Therefore, the ��-pH diagram with inclusion of Fe-As complexes is more meaningful to understand the As geochemistry in Fe-As-H2O system.
Fe-As complexes were not considered in previous ��-pH diagram mainly due to unreliability of thermodynamic data. Recently, MARINI and ACCORNERO [36] estimated the equilibrium constants and Gibbs free energies, which are in good accordance with other reports [37-39]. Accordingly, a new ��-pH diagram for Fe-As-H2O system was constructed based on the estimates. Figure 8(b) shows that Fe(III)-As(V) complexes shrink the stability field of H3AsO4 and FeAsO4(s). Moreover, Fe(II)-As(V) complexes occur in pH range of 0.5-3. The predominant field of Fe3(AsO4)2 is affected by Fe(II)-As(V) complexes. , and are all restricted at acidic pH conditions. Consequently, Fe-As(V) complexes are considerable in the As removal from smelting acidic wastewaters by coprecipitation with iron salts.
4 Conclusions
1) The cyclic voltammetry curves were tested in As(III)-containing sulfuric acid solutions (pH=1.0). It is shown that the over-potential of As(III) oxidation to As(V) is very high (about 0.9 V). Therefore, on-site peroxidation in acidic wastewater is challenging.
2) The UV-Vis spectra of a series Fe2(SO4)3- Na3AsO4-H2SO4-H2O solutions show that a new peak at about 240-300 nm appears as As(V) concentration increases. Corresponding Fe(III) species analyses indicate that Fe(III)-As(V) complexes are formed in the experimental solutions.
3) The ��-pH diagram for Fe-As-H2O system with inclusion of Fe-As(V) complexes were constructed and compared with the one with exclusion of Fe-As(V) complexes. The results illustrate that Fe(III)-As(V) complexes are stable under acidic conditions and shrink the stability field of H3AsO4 and FeAsO4(s).
References
[1] MANDAL B K, SUZUKI K T. Arsenic round the world: A review [J]. Talanta, 2002, 58(1): 201-235.
[2] CHAI Li-yuan, YUE Men-qing, YANG Jin-qin, WANG Qing-wei, LI Qing-zhu, LIU Hui. Formation of tooeleite and the role of direct removal of As(III) from high-arsenic acid wastewater [J]. Journal of Hazardous Materials, 2016, 320: 620-627.
[3] YANG W C, ZHAO N, ZHANG N, CHEN W, KAN A T, TOMSON M B. Time-dependent adsorption and resistant desorption of arsenic on magnetite nanoparticles: Kinetics and modeling [J]. Desalination and Water Treatment, 2012, 44:100-109.
[4] CHAI Li-yuan, SHI Mei-qing, LIANG Yan-jie, TANG Jing-jie, LI Qing-zhu. Behavior, distribution and environmental influence of arsenic in a typical lead smelter [J]. Journal of Central South University, 2015, 22(4):1276-1286.
[5] FILIPPOU D, DEMOPOULOS G P. Arsenic immobilization by controlled scorodite precipitation [J]. JOM, 1997,49(12): 52-55.
[6] CHAI L Y, CHEN Y N, YANG Z H. Kinetics and thermodynamics of arsenate and arsenite biosorption by pretreated spent grains [J]. Water Environment Research A: Research Publication of the Water Environment Federation, 2009, 81(9): 843-848.
[7] HARRIS B. The removal of arsenic from process solutions: Theory and industrial practice [J]. Hydrometallurgy, 2003, 2: 1889-1902.
[8] FUJITA T, TAGUCHI R, ABUMIYA M, MATSUMOTO M, SHIBATA E, NAKAMURA T. Novel atmospheric scorodite synthesis by oxidation of ferrous sulfate solution. Part I [J]. Hydrometallurgy, 2008, 90(2): 92-102.
[9] REDDY R G, RAMACHANDRAN V. Arsenic metallurgy [C]//TMS 2005 Annual Meeting. San Francisco, California, USA: Wiley, 2005.
[10] MIN Xiao-bo, LIAO Ying-ping, CHAI Li-yuan, YANG Zhi-hui, XIONG Shan, LIU Lin, LI Qing-zhu. Removal and stabilization of arsenic from anode slime by forming crystal scorodite [J]. Transactions of Nonferrous Metals Society of China, 2015, 25(4): 1298-1306.
[11] GONZALEZ-CONTRERAS P A. Bioscorodite: Biological crystallization of scorodite for arsenic removal [D]. Netherlands: Wageningen University, 2012.
[12] SINGHANIA S, WANG Q, FILIPPOU D, DEMOPOULOS G P. Temperature and seeding effects on the precipitation of scorodite from sulfate solutions under atmospheric-pressure conditions [J]. Metallurgical and Materials Transactions B, 2005, 36(3): 327-333.
[13] SINGHANIA S, WANG Q, FILIPPOU D, DEMOPOULOS G P. Acidity, valency and third-ion effects on the precipitation of scorodite from mixed sulfate solutions under atmospheric-pressure conditions [J]. Metallurgical and Materials Transactions B, 2006, 37(2): 189-197.
[14] PAKTUNC D, DUTRIZAC J, GERTSMAN V. Synthesis and phase transformations involving scorodite, ferric arsenate and arsenical ferrihydrite: Implications for arsenic mobility [J]. Geochimica et Cosmochimica Acta, 2008, 72(11): 2649-2672.
[15] YANG J, CHAI L, YUE M, LI Q. Complexation of arsenate with ferric ion in aqueous solutions [J]. RSC Advances, 2015, 5(126): 103936-103942.
[16] LONG H. A fundamental study of the acidic pressure oxidation of orpiment and pyrite at high temperature [D]. British Columbia: University of British Columbia, 2000.
[17] MASSCHELEYN P H, DELAUNE R D, PATRICK JR W H. Effect of redox potential and ph on arsenic speciation and solubility in a contaminated soil [J]. Environmental Science & Technology, 1991, 25(8): 1414-1419.
[18] PIECZABA E, SANAK-RYDLEWSKA S,  D. Removal of arsenic from aqueous solutions by the method of precipitate flotation [J]. Archives of Mining Sciences, 2005, 50(1): 131-142.
D. Removal of arsenic from aqueous solutions by the method of precipitate flotation [J]. Archives of Mining Sciences, 2005, 50(1): 131-142.
[19] YAZDI M R S, DARBAN A K. Effect of arsenic speciation on remediation of arsenic-contaminated soils and waters [C]//15th International Conference on Heavy Metals in the Environment (ICHMET). Gdansh, 2010: 492-495.
[20] WU H. An empirical estimation of standard entropy for some complex cations and the E-pH diagram of As-H2O system at elevated temperature [J]. Journal of Kunming University of Science and Technology, 1982, 3: 58-73.
[21] DEBUSSCHERE L, DEMESMAY C, ROCCA J L. Arsenic speciation by coupling capillary zone electrophoresis with mass spectrometry [J]. Chromatographia, 2000, 51(5-6): 262-268.
[22] GOUT R, POKROVSKI G, SCHOTT J, ZWICK A. Raman spectroscopic study of arsenic speciation in aqueous solutions up to 275 ��C [J]. Journal of Raman Spectroscopy, 1997, 28(9): 725-730.
[23] LOEHR T M, PLANE R A. Raman spectra and structures of arsenious acid and arsenites in aqueous solution [J]. Inorganic Chemistry, 1968, 7(9): 1708-1714.
[24]  J, PERSSON I, HERBERT R B. Hydration of arsenic oxyacid species [J]. Dalton Transactions, 2013, 42(5): 1364-1377.
J, PERSSON I, HERBERT R B. Hydration of arsenic oxyacid species [J]. Dalton Transactions, 2013, 42(5): 1364-1377.
[25] ROSSINI F D, WAGMAN D D, EVANS W H, ROSSINI F D, ROSSINI F D. Selected values of chemical thermodynamic properties [M]. Washington, DC: US Government Printing Office, 1952.
[26] SERGEYEVA E I, KHODAKOVSKIY I L. Physicochemical conditions of formation of native arsenic in hydrothermal deposits [J]. Geochemistry International, 1969: 846-859.
[27] Technical Note 270-7. SCHUMM R H, WAGMAN D D, EVANS W H, PARKER V B. Selected values of chemical thermodynamic properties [S]. National Bureau of Standards, 1973.
[28] WAGMAN D D, EVANS W H, PARKER V B, SCHUMM R H, HALOW I, BAILEY S M, CHURNEY K L, NUTTALL R L. Erratum: The NBS tables of chemical thermodynamic properties [J]. Journal of Physical and Chemical Reference Data, 1989,18(4): 1807-1812.
[29] BARD A J, PARSONS R, JORDAN J. Standard potentials in aqueous solution [M]. New York: CRC Press, 1985.
[30] SHOCK E L, HELGESON H C. Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: Correlation algorithms for ionic species and equation of state predictions to 5 kb and 1000 ��C [J]. Geochimica et Cosmochimica Acta, 1988, 52(8): 2009-2036.
[31] NORDSTROM D K, ARCHER D G. Arsenic thermodynamic data and environmental geochemistry [M]//Arsenic in Ground Water. Boston, MA, USA: Springer, 2003: 1-25.
[32] BU L, GU T, MA Y, CHEN C, TAN Y, XIE Q, YAO S. Enhanced cathodic preconcentration of As(0) at Au and Pt electrodes for anodic stripping voltammetry analysis of As(III) and As(V) [J]. The Journal of Physical Chemistry C, 2015, 119(21): 11400-11409.
[33] MARINI L, ACCORNERO M. Prediction of the thermodynamic properties of metal�Carsenate and metal�Carsenite aqueous complexes to high temperatures and pressures and some geological consequences [J]. Environmental Geology, 2007, 52(7): 1343-1363.
[34] PETTINE M, CAMPANELLA L, MILLERO F J. Arsenite oxidation by H2O2 in aqueous solutions [J]. Geochimica et Cosmochimica Acta, 1999, 63(18): 2727-2735.
[35] VINK B W. Stability relations of antimony and arsenic compounds in the light of revised and extended Eh-pH diagrams [J]. Chemical Geology, 1996, 130(1): 21-30.
[36] MARINI L, ACCORNERO M. Erratum to: Prediction of the thermodynamic properties of metal�Carsenate and metal�Carsenite aqueous complexes to high temperatures and pressures and some geological consequences [J]. Environmental Earth Sciences, 2010, 59(7): 1601-1606.
[37] LANGMUIR D, MAHONEY J, ROWSON J. Solubility products of amorphous ferric arsenate and crystalline scorodite (FeAsO4��2H2O) and their application to arsenic behavior in buried mine tailings [J]. Geochimica et Cosmochimica Acta, 2006, 70(12): 2942-2956.
[38] ROBINS R G. The stability and solubility of ferric arsenate: An update [EB/OL]. http://md1.csa.com/partners/viewrecord.php. 1990.
[39] WHITING K S. The thermodynamics and geochemistry of arsenic with application to subsurface waters at the sharon steel superfund site at Midvale, Utah [D]. Midvale: Colorado School of Mines, 1992.
[40] KNIGHT R J, SYLVA R N. Spectrophotometric investigation of iron(III) hydrolysis in light and heavy water at 25 ��C [J]. Journal of Inorganic and Nuclear Chemistry, 1975, 37(3): 779-783.
[41] HUG S J, CANONICA L, WEGELIN M, GECHTER D, von GUNTEN U. Solar oxidation and removal of arsenic at circumneutral pH in iron containing waters [J]. Environmental Science & Technology, 2001, 35(10): 2114-2121.
[42] WANG K L, JIA Y F. Effects of temperature and pH on the transformation of ferric arsenate to scorodite in acidic solution [J]. Advanced Materials Research, 2013, 726-731: 2165-2168.
[43] WELHAM N J, MALATT K A, VUKCEVIC S. The effect of solution speciation on iron�Csulphur�Carsenic�Cchloride systems at 298 K [J]. Hydrometallurgy, 2000, 57(3): 209-223.
�����1������Ԫ1,2��������1,2�������1,2
1. ���ϴ�ѧ ұ���뻷��ѧԺ����ɳ 410083��
2. ���ϴ�ѧ �����ؽ�����Ⱦ���ι��̼����о����ģ���ɳ 410083
ժ Ҫ��ұ�����Է�ˮ�����ȥ����һ��ʮ�ֽ��ȵĹ�����Ŀǰ��õķ������Ƚ�����������Ϊ����飬Ȼ���������ν��г���ȥ�������������ԭ��Ϊ����ѧ��̬�����о��������ȥ�������ش���ѭ��������������ɼ��ֹ��ȷ���չʵ�����о�������HSC��MINTEQ�����������۷������о������������������һ�������ת�Ʒ�Ӧ������ɢ���ƣ���pH 1.0��������Һ�У����������������λ�dz���(Լ0.9 V)�����⣬���Fe(III)- As(V)-H2SO4-H2Oϵ����Һ������-�ɼ������Լ�������̬����������Fe(III)-As(V)�����Ĵ��ڡ���ˣ����������о���Ԥ�������ѧ���ݣ�����Fe(III)��As(V)��������̬��pH�ķֲ�����ͼ�Ͱ���Fe-As���������͵�λ-pHͼ��
�ؼ��ʣ��飻��-��������λ-pHͼ����ѧ��̬�����Է�ˮ
(Edited by Bing YANG)
Foundation item: Projects (51304251, 51374237) supported by the National Natural Science Foundation of China; Project (201509050) supported by Special Program on Environmental Protection for Public Welfare, China
Corresponding author: Qing-zhu LI; Tel: +86-731-88830875; Fax: +86-731-88710171; E-mail: qingzhuli@csu.edu.cn
DOI: 10.1016/S1003-6326(17)60233-4

